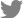Under natural conditions, plants are exposed to attacks by various pathogens, including fungi, bacteria, nematodes, viruses, and viroids. In particular, viruses are considered economically important because of their abundance in numerous crops (Akinyemi et al., 2016). Therefore, early virus identification is paramount to reduce crop losses and prevent viral disease spread.
Among the many plant species affected by viruses, Asian pear (Pyrus pyrifolia) is a widely cultivated and commercially important fruit crop in several regions worldwide, including Argentina, China, Italy, Korea, and Japan (Saito, 2016). Pear is one of the major pome fruit crops, with 10,000 ha of area under cultivation (Yoon et al., 2014). Over 3,000 pear cultivars are grown worldwide; however, a few common and economically important cultivars with different harvest times are grown in Korea (Elzebroek and Wind, 2008).
Pears are generally propagated via cloning and grafting to maintain cultivar traits; therefore, viruses and viroids that infect pear trees are readily transmitted. Major viruses infecting pear trees include apple chlorotic leaf spot virus (Trichovirus), apple stem grooving virus (ASGV; Capillovirus), and apple stem pitting virus (ASPV; Foveavirus) (Mahfoudhi et al., 2013). Although most viral infections of pear are asymptomatic, ASGV frequently causes black necrotic leaf spot disease symptoms, leading to significant economic losses in ‘Niitaka’ pear (Cho et al., 2010). In addition, multiple viruses and viroids simultaneously infect pear trees, potentially further reducing overall production.
At present, the detection of viruses and viroids typically involves the observation of viral disease symptoms, followed by the application of direct detection methods, such as enzyme-linked immunosorbent assay (ELISA), real-time polymerase chain reaction (RT-PCR), loop-mediated isothermal amplification, and recombinase polymerase amplification (Cho et al., 2010; Kim et al., 2018; Lu et al., 2018). However, these methods rely on prior information on the potential viruses and are only suitable for detecting known viruses and viroids and not for identifying novel viral pathogens.
approaches have furthered our understanding of viral genome variability and evolution as well as facilitated the identification of viral disease mechanisms, measurement of viral abundance and diversity, screening of asymptomatic viruses, detection of low viral titers in infected plants, and discovery of unknown viruses, thereby providing novel insights into the complexity of the viral biosphere. Several studies have previously used NGS to investigate viromes—defined as the collective genomes of viruses present in a specific host or environment—and documented the viromes of several crops (Akinyemi et al., 2016; Jo et al., 2017; Jo et al., 2018a, 2020). Therefore, high-throughput sequencing (HTS) can be integrated into the diagnostics and management of viral diseases, even when previously unknown viruses are involved. However, despite the increasing adverse effects of viruses on pear production, relatively little is known about viruses and viroids that infect this fruit crop. To this end, the aim of the present study was to employ metatranscriptomics for investigating RNA viruses infecting the most commercially important pear cultivar ‘Singo’ (SG) in Korea.
Materials and Methods
Plant materials
Pear leaves were collected from 35 orchards in five major pear-producing regions (Naju, Chonan, Namyangju, Ulsan, and Sangju) in Korea during the summer and fall seasons of 2017-2019 (Fig. 1A). The collection of these pear leaves was permitted by the owners of the orchards. Pear leaves with typical disease symptoms, such as deformation, chlorotic or necrotic leaf spots, and yellow veins, were collected from 100 SG trees (Fig. 1B). The collected SG leaf samples were pooled, ground in liquid nitrogen, and stored at ‒80°C.
RNA extraction and library preparation for RNA sequencing
Total RNA was extracted from the ground samples using the IQeasy Plus Plant RNA Extraction Mini Kit (iNtRON, Daejeon, Korea) according to the manufacturer’s instructions, and the quality and quantity of the extracted RNA samples were measured using Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) and gel electrophoresis. Libraries were prepared from rRNA-depleted RNA samples using the TruSeq Stranded Total RNA Library Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Briefly, following rRNA removal, the remaining RNA was chemically fragmented and random primed for reverse transcription. The average insert size of the library was 200 bp. Then, adapters were ligated, and PCR amplification was performed to selectively enrich the DNA fragments with adapters and amplify the library DNA. Agilent 2100 Bioanalyzer was used for quality control of the generated library, and the prepared library was sequenced by Macrogen Co. (Seoul, Korea) using HiSeq 4000 sequencer, generating 200-bp paired-end reads.
Transcriptome assembly and analysis
Before analyzing the transcriptomes of SG, the infection of several pear leaf samples by ASGV and ASPV was investigated by RT-PCR, which indicated that most pear trees were infected by either of the two viruses (data not shown). Transcriptome was assembled de novo using a workstation (two six-core CPUs and 256-GB RAM) with the Ubuntu 12.04.5 LTS operation system. Raw data obtained from the library were subjected to de novo transcriptome assembly. In addition, FASTQ files of the paired-end sequences from the library were subjected to de novo transcriptome assembly using gsAssembler v2.8 (Roche Diagnostics, Branford, CT, USA) with default parameters (Wylie et al., 2014). To identify viral sequences in the library, the assembled transcriptome contigs were compared with sequences in the viral reference database in NCBI (downloaded from https://www.ncbi.nlm.nih.gov/genome/viruses/) using MEGABLAST, which is markedly faster and more reliable for virus identification than other sequence similarity programs; the E-value cut-off was 1e-5. Following initial analysis, putative virus-associated contigs with mean sequence coverage values of <10-fold were discarded, and the remaining putative virus-associated contigs were compared with sequences in the NCBI NR (non-redundant proteins) database using tBLASTx. Finally, endogenous viruslike sequences were removed from the dataset, and virus-associated contigs were retained for further analyses.
Viral sequence mapping and genome assembly
To generate complete viral genomes, the pear transcriptome sequences were compared with a complete reference viral genome sequence using BLAST. Contigs with the greatest similarity to specific viruses were selected. The virus-associated contigs were mapped to the reference viral genome using Geneious v. 11.1.5 (Kearse et al., 2012), and consensus sequences for each mapping file were generated using a threshold of 95% identity.
Phylogenetic analysis
The complete genome sequences obtained through the preceding analyses were used to construct phylogenetic trees. The complete coat protein (CP) gene sequences of 20 ASGV isolates and 19 ASPV isolates were used for phylogenetic analyses. NCBI accession numbers of all viral genomes are indicated in the phylogenetic trees. Additional complete viral genome sequences were obtained for analysis by comparing the sequenced viral genomes with the ones in GenBank using BLASTN and by removing partial sequences from the results. Nucleotide sequences were aligned using CLUSTALW in BioEdit, and phylogenetic trees were generated using 1,000 bootstrap replicates based on either neighbor-joining or Kimura 2-parameter method in MEGA 5.05 (Tamura et al., 2011). Pairwise distances were calculated using the PASC algorithm, available from GenBank (Bao et al., 2012).
Calculation of viral abundance and copy number
To calculate viral abundance, MEGABLAST was performed against the sequenced reads associated with respective viruses. FASTQ files were converted to FASTA files using the FASTX-Toolkit (http://hannonlab.cshl.edu/fasx_toolkit/), and the resulting FASTA files were used for performing MEGABLAST. Copy number was calculated by multiplying the number of viral reads by 150 bp (average length of total reads) and then dividing the product by the total length (bp) of individual genomes (Jo et al., 2017).
Confirmation of the presence of identified viruses by RT-PCR
To confirm the presence of identified viruses in pear samples, RT-PCR was performed using virus-specific primer sets (Table 1). One-tube multiplex RT-PCR was performed in 20 μl reactions containing SuprimeScript RT-PCR Premixture (GeNet Bio, Daejeon, Korea), 2 μl RNA template, forward and reverse primers, and DEPC water. RT-PCR was conducted under the following conditions: 50°C for 30 min; 95°C for 5 min; 38 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 60 s; and 72°C for 5 min. The resulting amplification products were subjected to electrophoresis on 2% agarose gel in 1× TAE buffer, stained using ethidium bromide, and visualized under UV light. Product size was determined by comparison with a 100 bp DNA ladder (iNtRON).
Data availability
The raw dataset generated in the present study will be available, upon publication, in the NCBI Sequence Read Archive (SRA) repository under accession number SRR8466619. The viral genome sequences generated in the present study are deposited in GenBank with respective accession numbers.
Results
Library construction
Transcriptome datasets were generated from the library using paired-end sequencing. For viral metatranscriptomic analysis, transcriptome data were analyzed according to the experimental scheme (Fig. 1C). Raw RNA sequencing (RNA-Seq) data corresponding to the library are deposited in the SRA repository (Supplementary Table 1). A total of 6.7 Mb data were obtained from the SG library, corresponding to 78,624,272 reads.
Virus identification
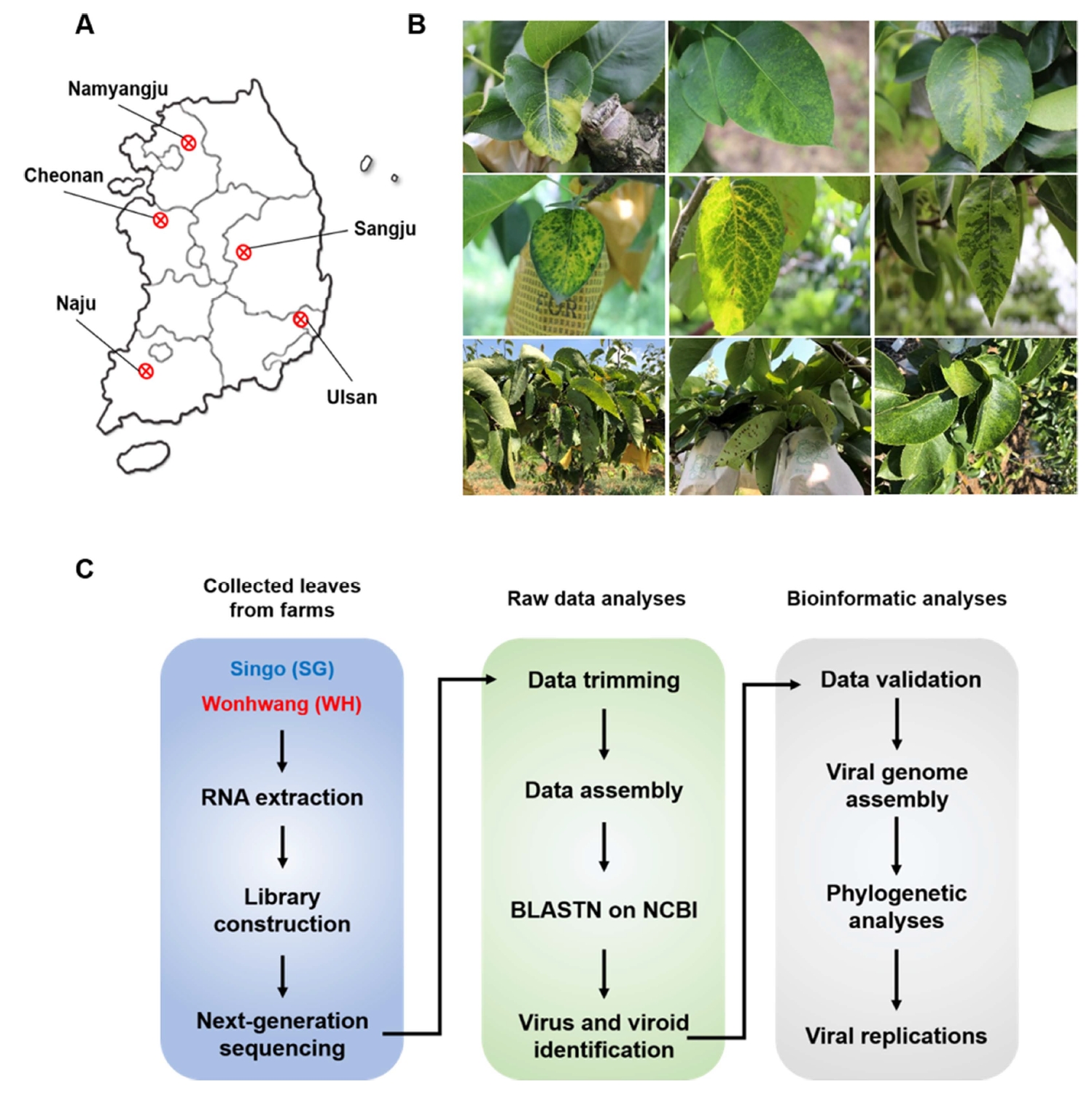
Raw reads from the library were de novo assembled using gsAssembler. The N50 values ranged from 993 to 1,126 bp (Supplementary Table 1). Moreover, 111,724 small (≥200 bp) and 35,725 large (≥500 bp) contigs were generated for the SG library (Fig. 2A).
The assembled contigs were compared with the sequences in the virus and viroid reference databases using BLAST. A total of 1,428 virus- and viroid-associated contigs were identified. After removing endogenous virus-associated sequences, 521 virus-associated contigs were identified.
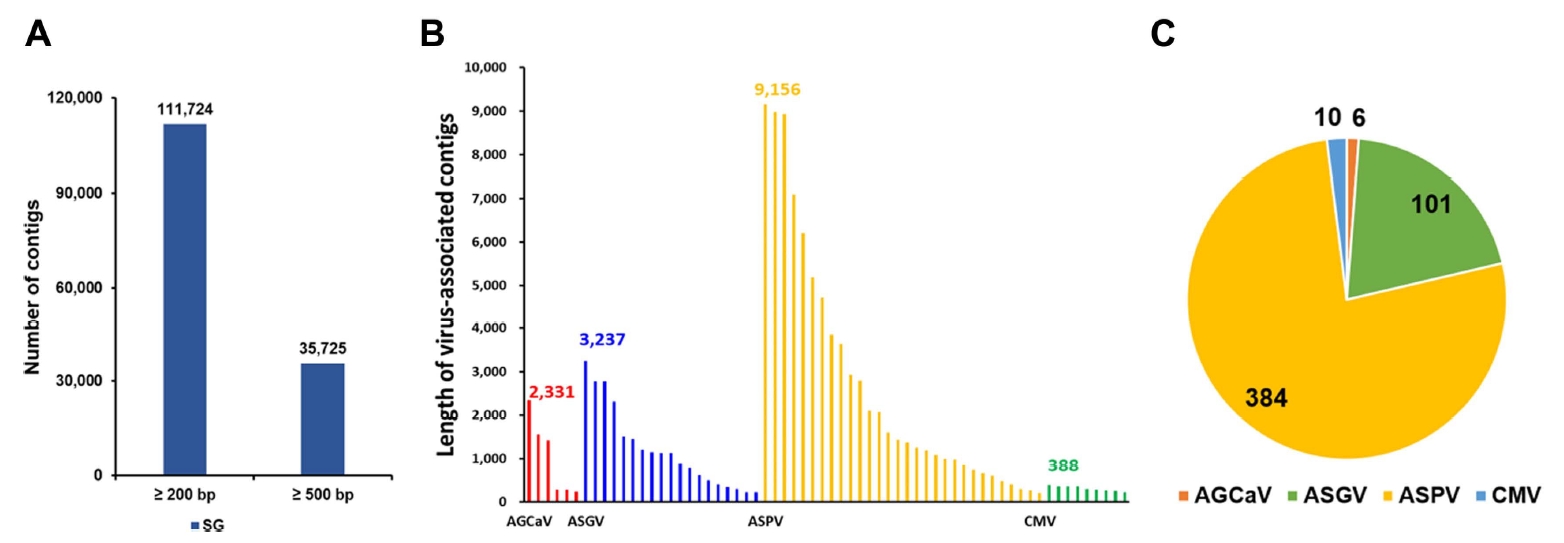
The virus-associated contigs from the dataset were first combined and then classified. The viruses identified from the transcriptomes included members of two virus families, namely Betaflexiviridae (511 contigs, 98.1%) and Bromoviridae (10 contigs, 1.9%). The length of virus-associated contigs ranged from 200 to 9,156 bp (Fig. 2B), and the largest seven contigs (>5,000 bp in length) likely represented the de novo assembled partial or nearly complete viral genomes. The virus-associated contigs from the SG library matched four reference viruses (Supplementary Table 2). In the SG library, ASPV-associated contigs (n = 384) were the most abundant, followed by ASGV- (n = 101), cucumber mosaic virus (CMV)- (n = 10), and apple green crinkle associated virus (AGCaV)-associated contigs (n = 6) (Fig. 2C).
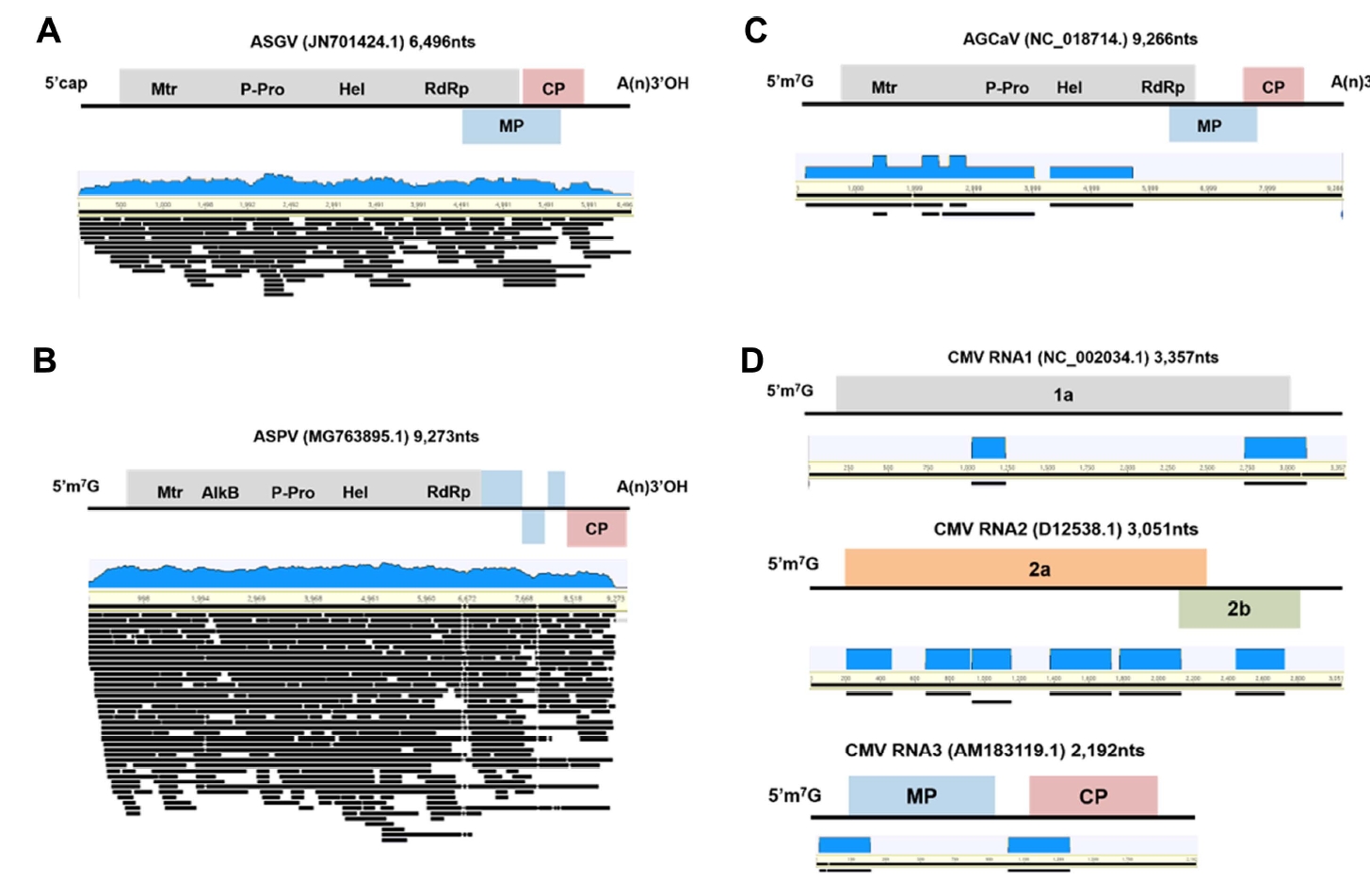
De novo assembly of viral genomes
To assemble complete or nearly complete viral genomes from the transcriptome data, the virus-associated contigs were mapped to the corresponding reference genomes (Fig. 3). Some ASGV- and ASPV-associated contigs covered the nearly complete genomes of the two viruses (Fig. 3A and B). Sizes of the virus-associated contigs ranged from 201 to 3,237 nt for ASGV and from 200 to 9,156 nt for ASPV (Supplementary Table 2). However, some sequence regions in genomes of the identified viruses were partially mapped to the virus-associated contigs (Fig. 3C and D). The size of AGCaV-associated contigs ranged from 236 to 2,331 nt and that of CMV-associated contigs ranged from 212 to 388 nt (Supplementary Table 2). In case of AGCaV, partial sequences were obtained from SG.
Phylogenetic relationships among identified viruses
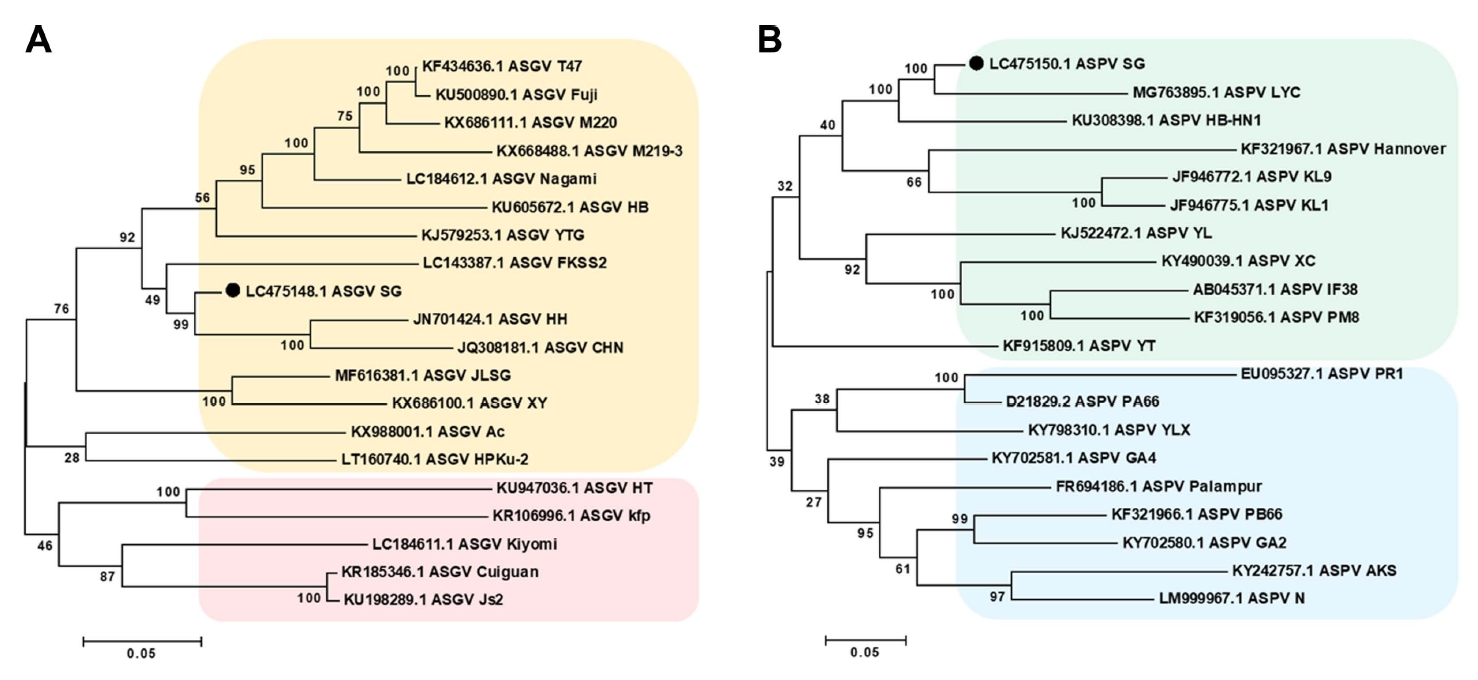
To determine the phylogenetic relationships among the identified viruses and their congeners, phylogenetic trees were constructed for each virus using the obtained nearly complete genomes based on the respective homologous sequence or viral genome. The assembled viral genomes were compared with sequences in the NT database using BLASTN. Owing to their high similarity between AGCaV and ASPV and incomplete assembly of AGCaV, phylogenetic analysis was performed with the complete sequences of ASGV isolates and ASPV isolates. The resulting phylogenetic tree indicated that the complete sequences included in the ASGV tree were separated into three clades. In the first clade, the ASGV isolate (accession no. LC475148) from the SG library was clustered with ASGV isolates derived from apple in China (Fuji, HB, T47, M220, and HH) (Fig. 4A). The second clade included ASGV isolates from citrus and pear in China (Cuiguan, JLSG, kiyomi, and Js2). The third clade included ASGV isolates from apple and pear in China (YTG, Ac, HT, and kfp). Similarly, complete sequences included in the ASPV tree, generated using a ASPV genome obtained in the present study and 18 additional genomes from GenBank (Fig. 4B), were separated into two clades. Interestingly, the ASPV isolate (accession no. LC475150) from the SG library was clustered with ASPV isolates from apple and pear in China.
Viral diversity
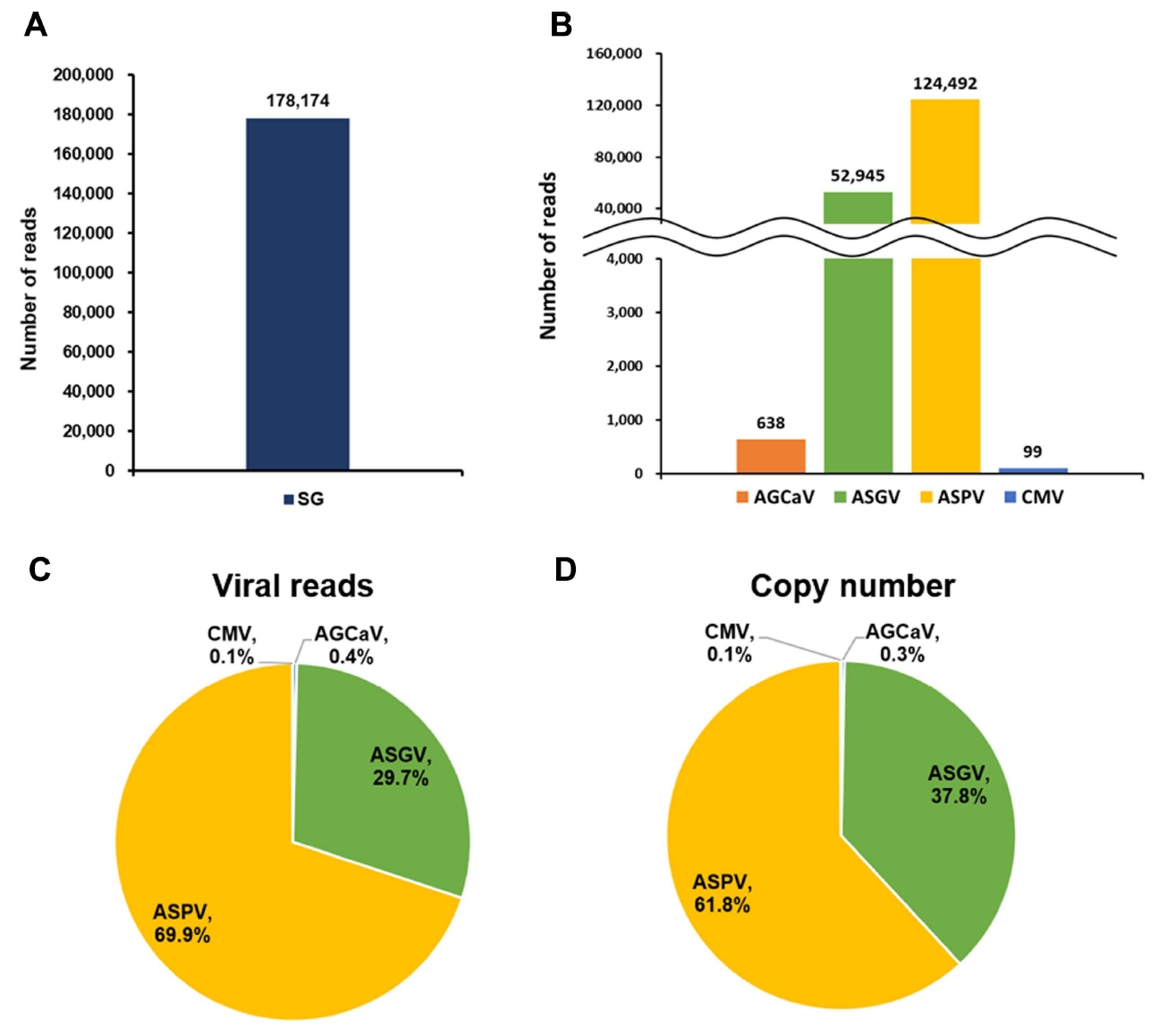
To calculate viral abundance, raw transcriptome sequence data were directly compared with reference genome sequences for the identified viruses using BLAST. The SG transcriptome (178,174 reads) contained numerous virus-associated reads (Fig. 5A). Furthermore, ASPV (124,492 reads) and ASGV (52,945 reads) were the dominant viruses in the SG library (Fig. 5B).
To determine the amount of individual viral RNA in the transcriptome based on read number, the number of total reads was divided by the number of virus-associated reads and multiplied by 151 bp. ASPV (69.9%) and ASGV (29.7%) were the dominant viruses in the SG library, followed by AGCaV (0.4%) and CMV (0.1%) (Fig. 5C).
Furthermore, the copy number of each identified virus was calculated using the number of virus-associated reads. Specifically, copy number was calculated by multiplying the number of individual virus- or viroid-associated reads by 151 bp and then dividing that value by the size (bp) of the corresponding viral genome. The results of the copy number were in accordance with those of viral abundance (Fig. 5D).
Confirmation of the presence of identified viruses by RT-PCR
RT-PCR with specific primers was performed to confirm the presence of identified viruses (Table 1). The presence of most identified viruses was confirmed using virus-specific primers designed based on a highly conserved region of the CP gene. However, it was difficult to differentiate between closely related AGCaV and ASPV. Several newly designed primers were tested, and specific primer pairs were selected to detect AGCaV. The RT-PCR products were purified and subjected to Sanger sequencing. BLAST results matched the identified viruses (Supplementary Fig. 1).
Discussion
Metatranscriptomic analysis of the virome of a major pear cultivar (SG) in Korea was performed. NGS is a recently emerged tool for virus detection and screening and has been used to discover numerous viruses, viral strains, and viral pathogens responsible for conditions of unknown etiologies (Jo et al., 2018a; Roossinck et al., 2015; Xu et al., 2019). Rapid virus discovery using NGS is considered a modern approach to prevent the outbreaks of novel viral diseases resulting from climate change, introduction via trade, and deterioration of natural ecosystems. Detection of viruses using classic methods, such as ELISA and RT-PCR, is often limited in terms of identifying novel virus species (Jeong et al., 2014; Ji et al., 2013). In addition to increasing the rate of virus discovery, NGS has been used to detect viruses and viroids in various fruit trees, including citrus, and peach (Jo et al., 2018b, 2020; Xu et al., 2019).
In the present study, viruses from pooled samples of infected pear leaves were identified and differentiated using a bioinformatics pipeline. Pooling is efficient for identifying viruses that infect plant species across a given geographical distribution. A single plant sample might yield useful information on the virome of a specific plant or population; however, such information is limited in its use for understanding the overall virome of pears in Korea. Pooling also reduces NGS cost. Therefore, pooled libraries and NGS techniques were used to identify viruses in the transcriptomes of a major pear cultivar. Contrary to the previous studies of plant virus metagenomics, the present study investigated RNA viromes of multiple pear plants, thereby facilitating the assessment of viral communities in a specific cultivar.
For virome analyses using NGS, sample preparation is a critical step. Most recent studies have prepared libraries of double-stranded RNAs, mRNAs, virus-derived small interfering RNAs, and plant rRNA-depleted total RNAs from the infected samples (Al Rwahnih et al., 2015; Donaire et al., 2009; Jo et al., 2018b) . Moreover, several recent studies have reviewed the applications of different sample preparation methods and proposed that the selection of nucleic acid extraction methods for HTS depends on diverse viral genome organizations and replication strategies (Wu et al., 2015). Consistent with other studies, massive viral RNA reads obtained from rRNA-depleted total RNA could be de novo assembled into viral genomes. In addition, length of the viral contigs is a crucial factor in the identification of viruses using RNA-Seq. Thus, BLAST results obtained for short contigs can provide misleading information for virus identification. Indeed, in the present study, BLAST analysis indicated the presence of two viruses, namely ASGV and ASPV, whose complete or nearly complete viral genome sequences were obtained.
Moreover, AGCaV and CMV were identified from the SG library, in addition to various viruses (e.g., american plum line pattern virus, citrus tatter leaf virus, and prunus necrotic ringspot virus) identified by BLAST analysis of short contigs. However, BLAST results for such short sequences sometimes lead to misidentification, even though partial sequences are derived from a single viral sequence (Jo et al., 2017). Although several studies have identified novel viruses of stone fruits using NGS (Jo et al., 2018a, 2020), no novel viruses or viroids of pear was identified in the present study. Nevertheless, the obtained information is useful for documenting the virosphere diversity of pear trees.
Nonetheless, there are some notable limitations to our employed pooling strategy, despite its cost-effectiveness. For instance, even though the procedure used a consensus viral genome sequence assembled from RNA-Seq data, viral sequence reads may be misaligned and recombination events may be misinterpreted due to pooling of multiple viral isolates (Schlötterer et al., 2015). Thus, pathological assays are required to investigate the concordance between the assembled viruses and sample pathogenicity using indicator woody plants such as pear trees showing susceptibility to pear viruses. In the present study, back inoculation of healthy pear trees based on Koch’s postulates couldn’t perform, since most pear viruses are transmitted through rooting, cutting, budding, and grafting, not by mechanical inoculation and are spread through infected propagules. Instead, we attempted to test back-inoculation to healthy herbaceous indicator plants such as Chenopodium quinoa, C. amaranticolor, Nicotiana benthamiana, N. tabacum, N. rustica, and N. glutinosa using mechanical inoculation. However, typical viral symptoms were not observed in the inoculated plants for 2 months (data not shown).
In the present study, phylogenetic analysis of viral genomes generated from the transcriptome data showed that ASGV and ASPV were closely related to isolates from China and Japan. Previous studies of phylogenetic relationships among ASPV isolates, analyzed using CP or triple gene block sequences, reported that the isolates were always clustered according to their hosts (Ma et al., 2016).
RNA viruses that infect plants typically exhibit high mutation rates, and many variants, which together comprise a quasispecies (Schneider and Roossinck, 2001), can often be observed in a single infected plant. In the present study, the quasispecies represented by pear tree-infecting viruses were examined by generating assembled virus sequences and analyzing the single-nucleotide variants (SNVs) of the two virus species. Because SNVs of the viruses were detected in library from pooled samples, the SNV information did not represent an individual virus. Even though several studies have reported the mutation rates of various viruses in plants, the present study detected relatively few SNVs (0-92) and relatively low mutation rates (0-1.42%), which suggests low levels of genetic diversity in the viromes of pear trees (Jo et al., 2017, 2018a).
In Korea, ASGV is highly prevalent, particularly in the SG cultivar, and it can lead to severe economic losses (Nam and Kim, 1994). Moreover, concomitant infection with multiple viruses is common in plants, and collectively, such viral infections can lead to substantial economic and yield losses (Katwal et al., 2016). To characterize virus populations infecting the SG pear cultivars, viral reads and copy numbers were calculated. Based on the transcriptome data, ASGV and ASPV were found to be the dominant viruses of the tested cultivar, although the copy number of ASPV was almost two-fold higher than that of ASGV.
Finally, RT-PCR was performed to confirm the presence of the identified viruses. ASGV, ASPV, AGCaV, and CMV were detected in SG samples. The present study demonstrated that HTS is a simple tool for identifying potential causative viruses prior to biological tests.
In summary, the present study describes the virome of SG, a major pear cultivar in Korea, and provides a comprehensive overview of the diversity of viral communities in this cultivar. HTS, bioinformatics and phylogenetic analyses, and viral RNA abundance in viromes can provide valuable information on the complexity of genetic variability of disease-causing viruses of pear. The present HTS-based metatranscriptomic analysis furthers genomic information on viruses infecting pear, enabling rapid response to emerging viral diseases of this fruit tree.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement1
Supplement1 Print
Print