Black rot, caused by Xanthomonas campestris pv. campestris (Xcc), is a transmissible disease of cruciferous crops (Williams, 1980). Xcc is a gram-negative bacterium that infects almost all cruciferous plants such as cabbage, mustard, radish, and Arabidopsis (Pammel, 1895). The pathogen usually invades from the leaf margin, resulting in “V”-shaped lesions that gradually expand inward to the veins. Subsequently, leaves and veins turn yellow or brown and the whole plant may become withered (Jensen et al., 2010; Fargier et al., 2011).
Black rot was firstly found on rutabagas and cabbages in the 1890s in the United States (Pammel, 1895), and has since then gradually spread all over the world and result in considerable crop failure every year. In some cases, black rot caused as high as 70% production loss. To prevent the disease, many strategies have been implemented including agricultural, chemicals, and physical measures. However, the utilization of resistant cultivars is the most ideal way for effective disease control.
The inheritance of plant resistance to black rot is complex because it is determined by the genotypes of both the pathogen and the host, i.e., the interaction between host and pathogen resulting in resistance or susceptibility (Vicente et al., 2001). Thus, breeding programs of black rot resistance in different regions should attach importance to local pathogen genetic structure. The traditional way to distinguish races of Xcc is based on their responses to a set of cruciferous cultivars. Till now, 11 races have been reported, with races 1 and 4 being the dominant races in Europe, Australia, and North America (Cruz et al., 2017; Fargier and Manceau, 2007; Vicente et al., 2001). Later, molecular methods, such as the 16S rRNA gene sequence variation, and amplified fragment length polymorphism (AFLP), were widely employed to uncover pathotypes because they are time-saving and reliable (Hauben et al., 1997; Restrepo et al., 1999). In the genomic era, genotyping technologies are frequently used for the identification of microorganisms, such as multilocus sequence genotyping (MLST) and repetitive DNA sequence-based PCR (rep-PCR) (Maiden et al., 1998; Versalovic et al., 1994). MLST is a genelevel approach based on nucleotide analysis that can provide phylogenetic relationships and genomic relatedness of the pathogens, and has currently become an effective and powerful method for genetic structure analyses and phylogenetic investigations of microbial strains such as Yersinia pseudotuberculosis and Streptococcus pneumonia (Achtman et al., 1999; Enright an Spratt, 1998). Also, rep-PCR is a fingerprint analysis method that reveals the differences between the genomes by amplifying and analyzing the repetitive sequences that are widely distributed in the bacterial genomes (Versalovic et al., 1994). It has been used to assess the diversity and phylogenetic structure of bacteria such as Lactobacillus and Pseudomonas (Gao et al., 2016; Hyytiä-Trees et al., 1999). Moreover, MLST analysis and rep-PCR are frequently used in combination to reveal genetic diversity and new pathotypes of microbial groups. For example, to investigate the pathogenic diversity of Xcc strains in India, genetic characterization of different strains was conducted using rep-PCR and MLST (Rathaur et al., 2015). The results of rep-PCR showed the highest correlation (70%) with those obtained with MLST performed with housekeeping genes (Popović et al., 2019).
China produces the largest amount (over 40%) of cruciferous crops around the world, and black rot was first reported in the northern area in the 1950s and has since then spread across the country, causing considerable yield and quality reduction to the cruciferous crop production. In recent years, due to inappropriate farming methods and climate change, the damage caused by black rot is gradually increasing (Lv et al., 2020; Zhang et al., 2011). Moreover, black rot poses a great threat to cole crops due to the lack of resistant cultivars, especially to cabbage, an important leafy vegetable that provides abundant nutrients as well as health-promoting substances for humans worldwide. Identification of the pathogen genetic diversity, population structure, and virulence is a fundamental work for prevention against black rot. Although some studies on the pathogenicity of Xcc have been conducted in China (Lu et al., 2008), the population diversity of Xcc is still largely unclear. In this study, we collected 50 Xcc strains, including 39 strains isolated from black rot-infected cabbages of 20 different areas of China, as well as seven type races derived from other countries (USA, UK, Australia, and Portugal). These Xcc strains were analyzed using MLST, rep-PCR, and inoculation tests to reveal their genetic diversity, population structure, and virulence. The results should be helpful to both cabbage resistance breeding and Xcc pathogenicity studies.
Materials and Methods
Collection of Xcc strains and DNA preparation
Leaves of 39 cabbage samples showing typical black rot symptoms were collected in 2016-2019 from 20 cities or provinces across China. Races 1-6 strains were provided by University of Warwick, UK. Strains race 9 and XCBS were provided by Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences (IVF-CAAS), and XCC1, XCC2 and XCC4 were provided by Beijing Academy of Agriculture and Forestry Sciences (BAAFS), Beijing, China. The details about the strains used in this study are shown in Table 1 and Supplementary Fig. 1. Isolates from leaf tissues were cultured on complete mediums (with agar) for 16 hours, and a single bacterial colony of the cultures was selected for reproduction in complete mediums (without agar) for 16 h at 28°C on a rotatory shaker. The isolates were validated to be Xcc by 16S ribosomal RNA amplification (Jiang et al., 2006), morphology characterization using microscopic examination, and pathogenicity test to fulfill the Koch’s postulates. The Xcc strains were then incubated and subjected to bacterial genomic DNA extraction using an extraction kit following the manufacturer’s instructions (Tiangen Biotech Co., Ltd., Beijing, China). DNA quantity was measured by 1% gel agarose electrophoresis and assayed based on the ultra-micro spectrophotometer (Biochrom Co., Ltd., Cambridge, UK). The concentration of all DNA was diluted to 50 ng/μl with double-distilled water.
Sequence assays of housekeeping gene fragments
Four genes, including three housekeeping genes, atpD (ATP synthase subunit beta), gyrB (DNA gyrase subunit B), rpoD (RNA polymerase sigma-70 factor), and the gene fyuA (TonB-dependent receptor) encoding a transmembrane protein were amplified by primers described by (Fargier et al., 2011). All reactions were performed in a final volume of 20 μl containing 10 μl of 2× Phanta Max Master Mix (Vazyme Biotech Co., Ltd., Nanjing, China), 1 μl of each forward and reverse primer (10 pmol/μl), 1 μl of DNA (50 ng/μl), and 7 μl of double-distilled water. PCR reactions were carried out with cycling conditions as follows: an initial denaturation of 94°C for 3 min, 35 cycles of 94°C for 50 s, varied annealing temperature for 50 s, and 72°C for 1 min, and a final extension of 72°C for 7 min (Table 2). Amplification was confirmed on 1.2% agarose gels stained with the nucleic acid dye (Vazyme Biotech Co., Ltd.) and visualized on the gel image analyzing system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Sequencing was performed using ABI 3730 xl DNA Analyzers (Applied Biosystems, Foster City, CA, USA).
Sequence alignment and data analysis
Sequences were initially analyzed and corrected using DNAMAN v. 6.0 (Lynnon Biosoft Co., Quebec, Canada), and then they were compared using the strain ATCC33913 at GenBank (https://www.ncbi.nlm.nih.gov/) to determine the accuracy. The sequencing results of these 50 strains were aligned and trimmed in GenBank with the ClustalW program in MEGA v. 6.0 (Tamura et al., 2013), and the gene length, numbers, and polymorphism sites of alleles were redefined based on the findings of previous studies (Bella et al., 2019; Fargier et al., 2011). Each gene sequence was identified as a new allele and given an allele number if the sequence displayed diversity at any nucleotide site, and then the allelic profile (AP) was defined by combining the allele numbers for each strain. Different APs indicated distinct sequence types. According to the concatenated sequences of four housekeeping genes of each strain, the phylogenetic relationship and the neighbour-joining trees were constructed with cluster analysis by the unweighted pair-group and the arithmetic average. To uncover population genetics, nucleotide and haplotype diversity analysis were performed using DNA polymorphisms in DNASP v. 5.0 (Librado and Rozas, 2009). Every unique pair of sequences and the identity score for each pair of sequences were constructed using the NW algorithms implemented in the ClustalW alignment using SDT v. 1.2 (Muhire et al., 2014). The NEIGHBOUR component of PHYLIP was used to generate a sequence phylogenetic tree under the rooted neighbour-joining condition, and the sequences were ordered by the degree of evolutionary relatedness according to the score calculated by the tree rearrangement. Finally, pairwise identity plots and colour-coded distance matrices with concatenated sequences of the 50 strains of Xcc were generated using SDT v. 1.2.
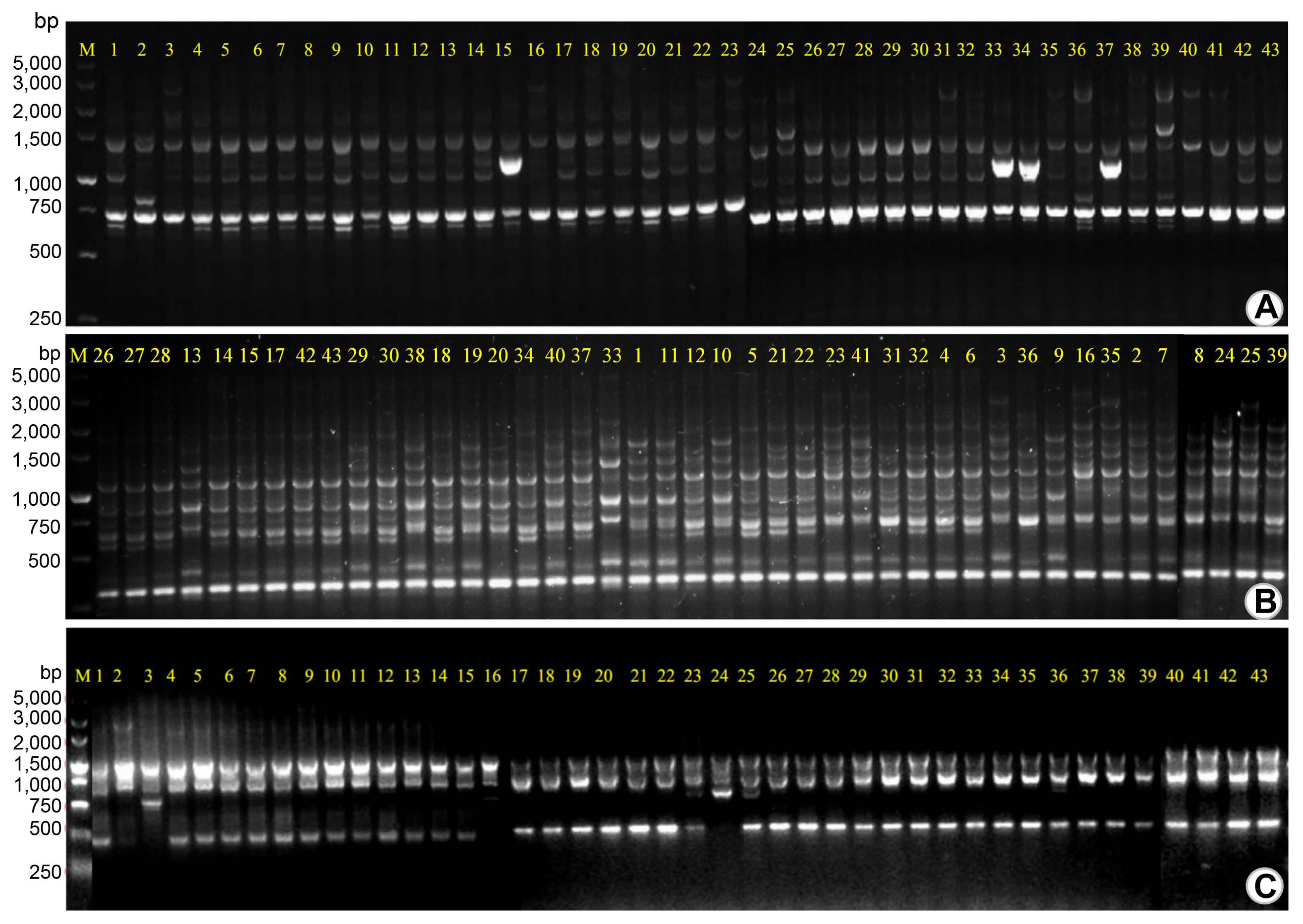
Rep-PCR amplification and polymorphism statistics
Genome characterization of all strains was performed by using three rep-PCR, namely, REP PCR (repetitive extragenic palindromic), ERIC PCR (enterobacterial repetitive intergenic consensus), and BOX PCR (BOX element sequences) (Martin et al. 1992; Versalovic et al., 1991). The rep-PCR primers described by Louws et al. (1994) included three pairs of primers for REP, ERIC, and BOX. The PCR amplification volumes and conditions were adjusted appropriately according to Rademaker and De Bruijn (1997). A 25 μl of reaction mixture for PCR amplification contains 12.5 μl of 2× Phanta Max Master Mix (Vazyme), 3.5 μl of DNA (50 ng/μl), 1 μl of forward primer (10 pmol/μl), 1 μl of reverse primer (10 pmol/μl), and 5 μl of double-distilled water. The PCR reaction conditions consisted of 7 min denaturation at 94°C followed by 33 cycles of 94°C for 1 min, annealing at 40°C, 52°C, or 53°C for 1 min for REP, ERIC, and BOX primer sets, respectively, and 7 min extension at 65°C, with a final extension of 15 min at 65°C (Table 2). All PCR amplicons were confirmed on 1.5% agarose gels and visualized on the gel image analyzing system (Bio-Rad Laboratories, Inc.). The electrophoresis pattern of “0/1” amplified DNA fragments was determined by counting identifiable amplicons recorded as “1” and missing amplicons recorded as “0.” The “0/1” electrophoretic patterns were transferred into an EXCEL sheet (Microsoft Co., Redmond, WA, USA). The data were standardized according to the requirements of the clustering analysis software NTSYS-pc v. 2.1 (Exeter Software, Setauket, NY, USA), and the similarity matrix between the strains was calculated based on the band type statistical results of rep-PCR. The clustering tree was generated using the treeplot module, and finally, the correlation coefficient of the trees was calculated to determine whether the clustering was reasonable.
Virulence tests
Race 1, race 4, and 10 representative Xcc isolates collected from different Provinces from China were tested for their virulence by artificial inoculation to three brassica hosts, i.e., XGBH (Chinese kale, B. oleracea var. alboglabra), WJ006 (turnip, B. rapa), and 87-534 (heading cabbage, B. oleracea var. capitata), which were all provided by IVF-CAAS. Simply, four true leaves of 30-day-old plants were inoculated using toothpicks that had been dipped in a suspension from a 16-hour-old culture of Xcc grown on a complete medium at 28°C (Kong et al., 2021). The disease reaction was surveyed on 14th day after inoculation. Disease grade for each inoculated leaf was determined using the following scale: 0, healthy leaf with no lesion; 1, slight necrosis or chlorosis surrounding the infection point, with the area being less than 5% of the leaf; 3, small lesion, with the area accounting for 5-15% of the leaf; 5, small V-shaped lesion with slight black veins, with the area accounting for 15-30% of the leaf; 7, typical V-shaped lesion with black veins, with the area accounting for 30-50% of the leaf; 9, the large lesion covered more than 50% of the leaf. The disease index was calculated as follows: disease index (DI) = ∑(disease rating scale × the number of leaves in a scale) × 100/(the total number of tested leaves × 9).
Results
Sequence assay of housekeeping genes
The primers designed by Fargier et al. (2011) were used to amplify the target genes. After the PCR products subjected to agarose gel electrophoresis, the fragment size of each housekeeping gene was consistent with the expected results and met the sequencing requirements for the single band. To ensure the accuracy of the sequencing results, the bidirectional DNA sequencing method was used to detect PCR products. After splicing the forward sequences and reverse sequences, suspicious single nucleotide polymorphisms (SNPs) were repeatedly confirmed by combining the figure of the sequencing peaks (Fargier et al., 2011). The sequences of each of the four loci were aligned with the 48 allele sequences from Fargier et al. (2011) and six allele sequences from Bella et al. (2019) published in GenBank on X. campestris crucifer-attacking pathovars. Sequence alignment of each of the four loci showed no gap and insertion events and only SNP difference in the sequences. All the sequences of each housekeeping gene were aligned and the two ends of each alignment were trimmed to 648 nt for atpD, 771 nt for fyuA, 705 nt for gyrB, and 807 nt for rpoD, respectively (Table 3). The sequences of four housekeeping genes of 50 Xcc strains were concatenated based on their order in the genome: rpoD followed by fyuA, atpD, and gyrB. The final sequence of the housekeeping genes for each strain of Xcc was 2,931 nt (Table 3). The analyzed sequences of all strains are placed in the Supplementary Fig. 2. All allele sequences of X. campestris strains obtained in this study were deposited in the NCBI GenBank database under the accession numbers MW429453 to MW429459 (atpD fragment), MW429460 to MW429465 (fyuA fragment), MW429466 to MW429470 (gyrB fragment), MW429471 to MW429475 (rpoD fragment). By BLAST analysis (http://bacteria.ensembl.org/index.html/BLAST), the identity of these housekeeping gene fragment sequences and strain ATCC 33913 of Xcc was higher than 97%.
AP analysis
DNA sequences from each of the four loci that differed from each other by one or more polymorphisms were assigned an allele number, and the combination of allele numbers for each strain defined the AP. Fifty strains of Xcc were classified into 12 different genotypes (Table 1). AP1 had the largest number of strains, including 32 strains collected from 16 regions (Chongqing, Beijing, Shandong, Shaanxi, Henan, Hubei, Zhejiang, Gansu, Hebei, Neimenggu, Jiangxi, and Shanxi), and was the dominant sequence type. Four strains of Xcc were grouped into AP8, including race 1, XCC2, XCC4 and XCTY, and only XCTY was isolated from China. Two APs (AP6 and 7) each contained three strains, whereas the remaining APs (AP2-5, 7, 9-12) were unique with only one Xcc strain. Race 2 was grouped into AP9, and races 3 and 9 were divided into AP7. Races 4, 5 and 6 were classified into AP10, 11 and 12, respectively. The results showed that AP1 was a large and unique group (alleles 1-1-1-1 for atpD, fyuA, gyrB and rpoD, respectively), of which the gene sequences showed differences with all type races in the study. Thus, AP1 might represent a new pathotype based on the comparison with the races 1-6 and 9 at the genetic level.
Genetic diversity of MLST
In this study, DNASP v. 5.0 was used to calculate the number of alleles, polymorphisms, nucleotide diversity (π), and haplotype diversity (Hd) of four housekeeping genes of 50 Xcc strains. The fragments of these gene sequences ranged in length from 648 to 807 bp, and the total length of four housekeeping gene concatenated sequences was 2,931 bp. The number of alleles reflected the genetic diversity of the housekeeping genes, and four housekeeping alleles had numbers between five to seven, containing five alleles of the rpoD loci, seven alleles of the atpD loci, six alleles of the fyuA loci and five alleles of the gyrB loci (Table 3). In terms of the number of alleles, the fragment of gene gyrB was relatively stable with the smallest change in genetic diversity. The haplotype diversity of the atpD, fyuA, gyrB, and rpoD locus was 0.572, 0.385, 0.410, and 0.517, and the variance of the haplotype diversity was 0.00585, 0.00711, 0.00669, and 0.00601, respectively. Polymorphic sites of four housekeeping genes varied from 13 for the least polymorphic locus (gyrB) to 51 for the most polymorphic locus (rpoD), and the total concatenated sequences included 108 polymorphic sites (Table 3). Nucleotide diversity reflected the polymorphic mutation of the different locus, and it varied between zero to one. The nucleotide polymorphic variable values of four genes in this study were less than 0.02 and therefore showed low allelic variation.
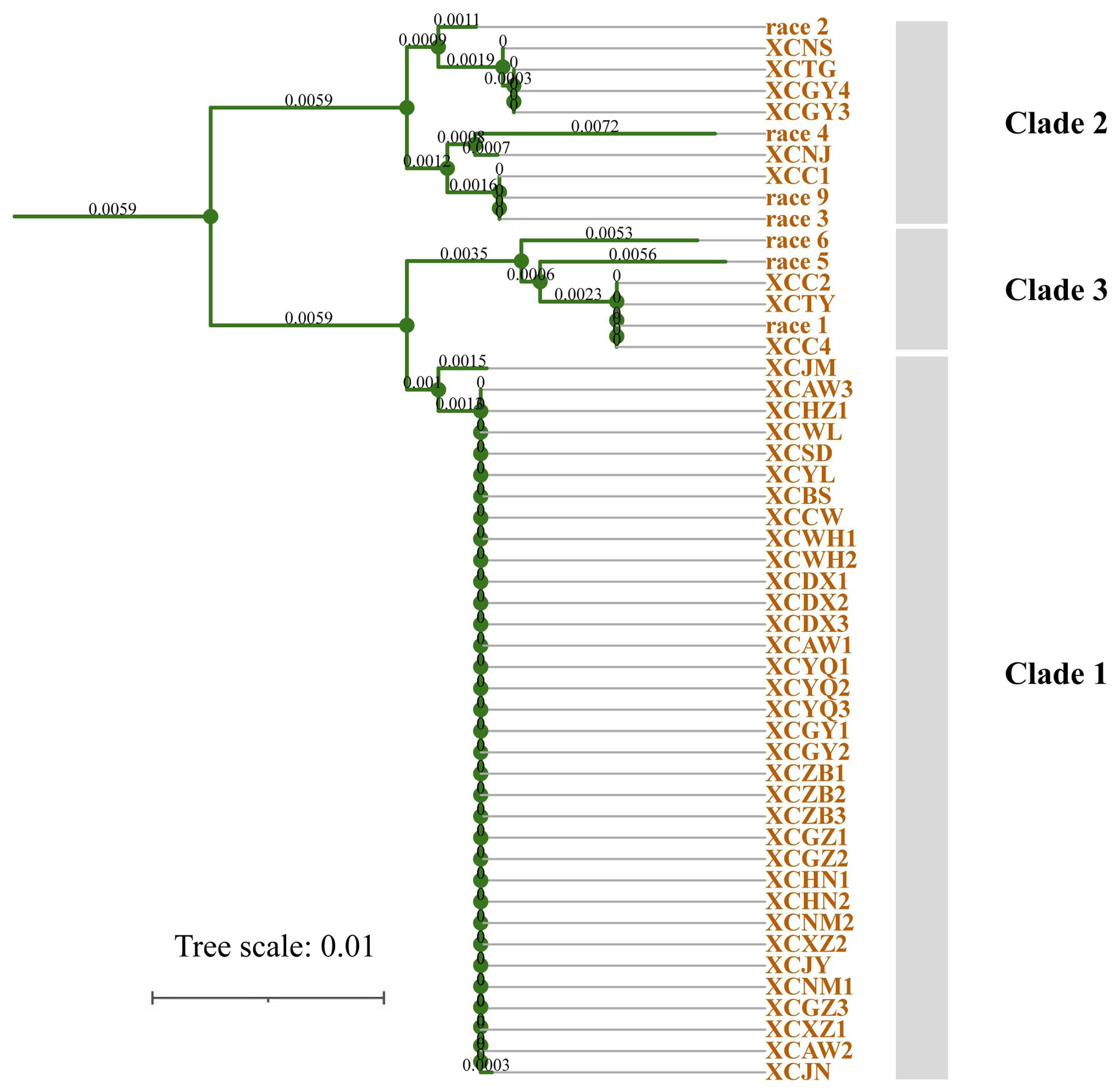
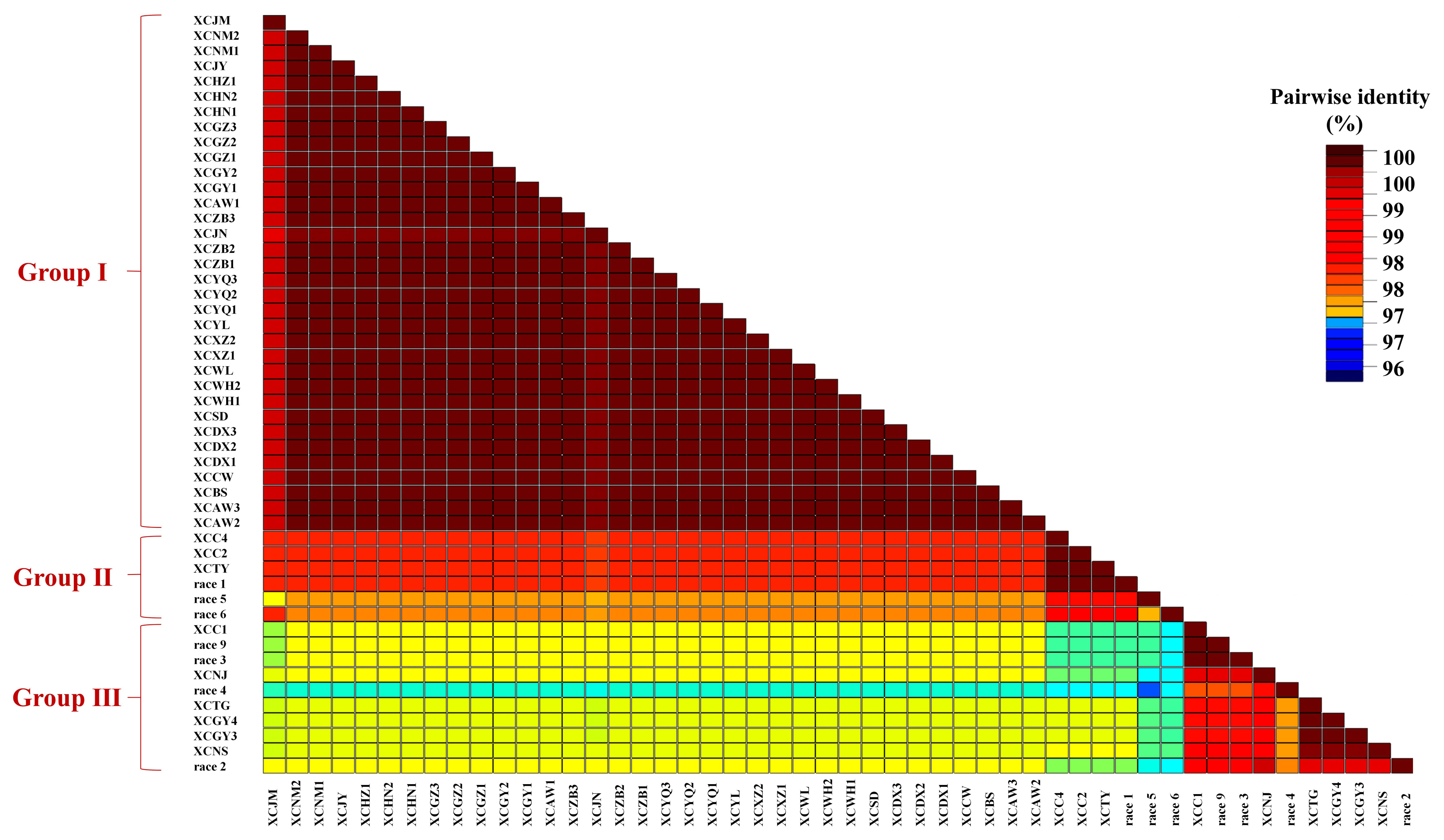
Phylogenetic analysis was performed for 2,931 concatenated sequences of individual genes of 50 Xcc strains, and the Xcc samples were distributed in three clades in the phylogenetic relationship and the neighbour-joining trees constructed with cluster analysis (Fig. 1). Clade 1 contained three phylogenetic subgroups, including strain XCJM from Hubei, XCJN from Shandong and the other 32 strains from 16 regions, which showed only a single nucleotide difference between strain XCJN and the other 32 strains. The other three subgroups, including race 1, race 5, race 6 and three other strains (XCC2, XCC4, and XCTY), were gathered together in Clade 2. Clade 3 contained six subgroups, among which races 2, 3, 4, and 9 were divided into three different branches. However, none of the type races was assigned to Clade 1 according to the phylogenetic relationship. Six of seven races were distributed in different subgroups, while races 3 and 9 were divided into the same subgroup (Fig. 1). Based on the concatenated sequences of four housekeeping genes, the pairwise identity of the 50 strains of Xcc was 97-100%, and the strains were distributed in three genetic groups based on 97% as a threshold (Fig. 2). Group I was identical to the distribution of the samples or strains of the pathogen in the above-described Clade 1 phylogeny. The lowest sequence identity was 97% between strain race 4 and strain race 5 (group III) (Fig. 2). Sequence identity varied between 99.9% to 100% among the 34 Xcc strains of Group I.
Genetic variation of rep-PCR
To determine the genetic relationship and variation characteristics of these Xcc strains, three sets of primers (ERIC, BOX, and REP primers) were used to amplify the DNA repetitive sequences extracted from these strains, and clear electrophoretic patterns were obtained. The patterns from the rep-PCR of all strains consisted of 6 to 15 fragments ranging in size from 0.4 to 5.0 kb (Fig. 3). The total number of DNA amplified bands was 31, among which the ERIC primers amplified 15 bands, BOX primers amplified 6 bands and REP primers amplified 10 bands, and the polymorphism ratio of DNA bands amplified by these primers was 80%, 93.3%, and 83.3%, respectively. According to the results of two repeated experiments, the number and length of the bands of some strains were significantly different and indicated that these strains might have variation and are polymorphic at the DNA level. As being shown in Fig. 3, some strains from the same place also showed band differences, indicating that different pathogenic strains could be distinguished according to the rep-PCR result.
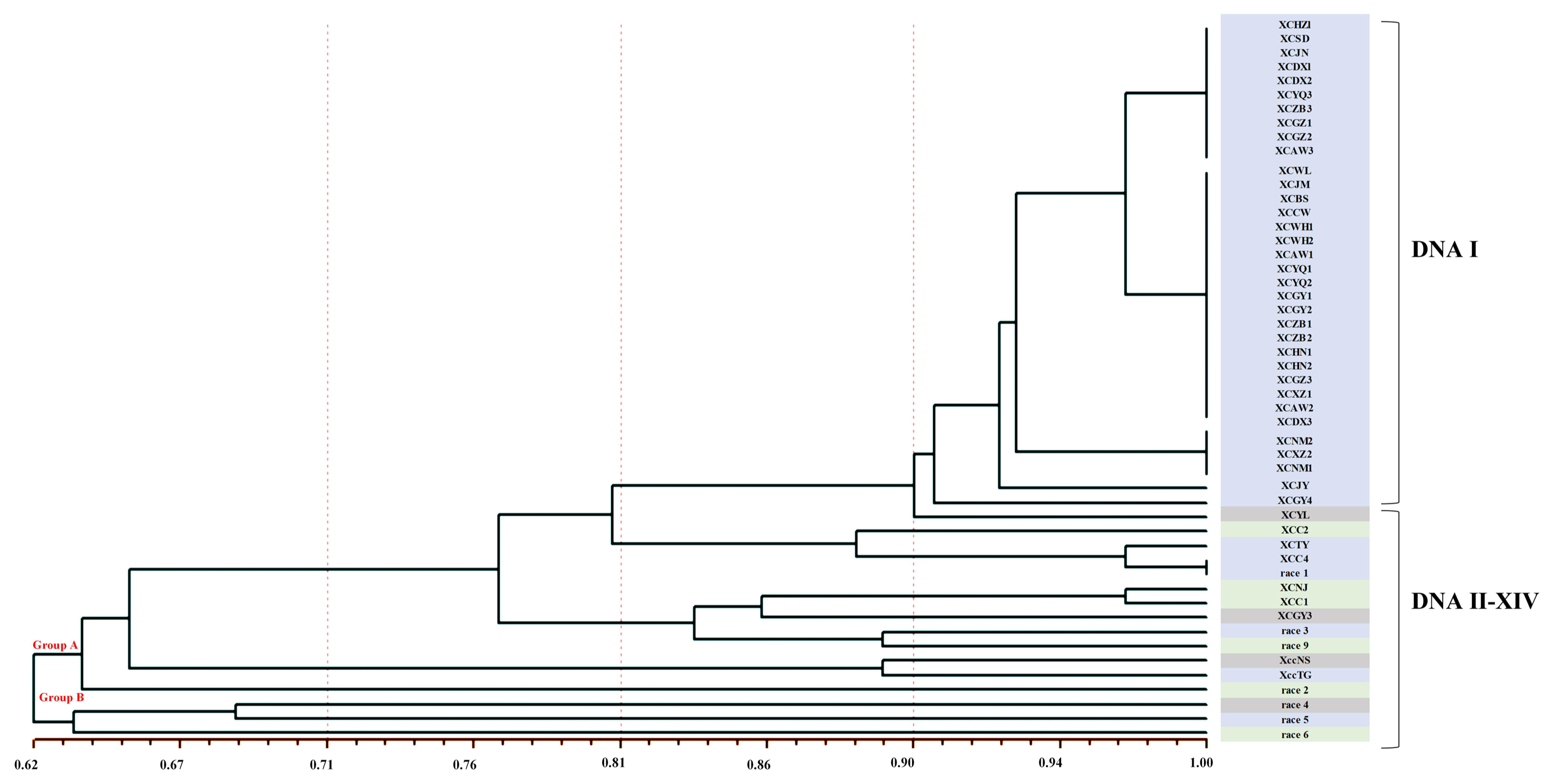
Clustering and phylogenetic analysis
To study the phylogenetic relationship of the pathogenic varieties of Xcc, the similarity module in the NTSYS-pc v. 2.1 was used to obtain the similarity matrix. In the SAHN module, clustering analysis was performed for all Xcc strain samples. Finally, the cophenetic correlation test and tree plot were performed (Fig. 4). The results showed that at the similarity level of 0.62, these strains collected from different areas could be divided into A and B genetic groups, including 47 and three strains, respectively. When the similarity coefficient was 0.90 as the threshold, Group A could be further divided into 10 subgroups. From the similarity level of 0.90 in the cluster map, DNA I was the largest cluster, including 34 strains collected from 17 cities or provinces including Zhejiang, Shandong, Gansu, Beijing, Hebei, Jiangxi, Chongqing, Hubei, Shaanxi, Henan, and Neimenggu. All strains of DNA I might be assumed to be the same pathotype and the main causal pathogen of black rot in most areas in China. In addition, several unique strains were found in the cluster map, which was divided into a separate subgroup including XCTG from Shanxi, XCNJ from Jiangsu, XCGY3 from Hebei, and XCNS from Guangdong (Fig. 4). According to the two methods, the main pathotype in China distinguished from the seven race strains originated from other regions including the USA, UK, Australia and Portugal. By combining the results of MLST and rep-PCR, the genotype of as many as 31 strains belonged to both AP1 and DNA I, indicating that the two different methods showed a high correlation due to the 90% similarity between AP1 and DNA I.
Virulence tests
All three brassica hosts showed clear phenotypic symptoms, after being inoculated with 12 representative Xcc strains, including strains type race 1 and 4, isolates XCNM1, XCTY, XCGY3, XCJY, XCNJ, XCGZ1, XCWL, XCYL, XCCW, and XCDX1 collected from Neimenggu, Shanxi, Hebei, Hubei, Jiangsu, Jiangxi, Chongqing, Shaanxi, Shandong, and Gansu provinces, respectively. These Xcc strains were pathogenic to different hosts, but showed significant differences in virulence (Table 4). Brassica hosts XGBH and 87-534 developed symptoms of high susceptibility to all strains, while WJ006 showed different degrees of susceptibility to these strains. According to the necrotic leaf lesions symptoms on inbred line WJ0006, seven strains XCJY, XCNM1, XCGZ1, XCWL, XCYL, XCCW, XCDX1, divided into Clade 1 by genotyping showed low-pathogenicity (DI < 10), two Xcc strains XCGY3 and XCNJ grouped into Clade 2 displayed medium-pathogenicity (40 < DI < 45), and strains XCTY and race 1 belonged to Clade 3 exhibited high pathogenicity (59 < DI < 78). Isolate XCTY obtained from cabbage not only caused the largest lesions on leaves of inbred Line WJ006 (B. rapa), with the DI of 77.8 ± 3.6, but also showed a broader spectrum of pathogenicity compared with the world mainstream type race 1, since it caused high susceptibility symptoms on all of the tested hosts. Moreover, the pathogenicity differentiation of Xcc strains collected from 10 distinct regions indicated that the dominant pathogenic type in China differs from the mainstream races 1 and 4 in the world.
Discussion
Black rot first occurred in Northern China in the late 1950s, and it has become a severe disease in recent years, distributed widely in main cole crop planting regions of China. Characterization of the pathogen is a vital step for disease prevention. More recently, to reveal the pathogenetic types, various high-efficiency molecular techniques have been developed, such as MLST and rep-PCR (Garces et al., 2014; Gonçalves and Rosato, 2002; Rademaker et al., 2000).
MLST was a molecular typing method performed directly at the genetic level to characterize pathogens and the MLST system is currently widely used to study bacterial population evolution, epidemiology, and pathogenicity (Maiden et al., 1998; Ntambo et al., 2019; Rathaur et al., 2015; Urwin and Maiden, 2003). The major advantages of the analytic method include the high resolution of MLST technology, the ability to record changes at the bacterial gene level, and the ease of obtaining materials for MLST detection. Previously, some studies have focused on the identification and analysis of Xanthomonas strains using the MLST system (Ah-You et al., 2009; Bella et al., 2019; Rathaur et al., 2015). A study based on the analysis of the sequence fragments of eight housekeeping genes suggested a high genetic diversity of pathovars of X. campestris derived from gene recombination and point mutation accumulation (Fargier et al., 2011). Molecular markers were designed to amplify 434-697 bp fragments of eight housekeeping genes. The number of polymorphic nucleotide sites varied from 8 to 71 and the number of alleles ranged from 6 to 15. In our study, to evaluate the variation in housekeeping genes, we used the primers reported by (Fargier et al., 2011), and the gene sequences were trimmed to 648 to 807 bp to obtain additional polymorphic sites. Our findings indicated that SNPs can be obtained by appropriately increasing the length of the gene fragment. Based on the APs of four housekeeping genes obtained in the analysis, we found that the bacterial populations observed in China were as variable as the strains collected in Europe (Bella et al., 2019; Lema et al., 2011; Rathaur et al., 2015). Our results showed that 12 different genotypes were identified among the 50 strains, and AP1 was the largest clade and the most widespread among the Xcc strains. Although some different APs were detected in different geographical areas, 32 strains collected from diverse regions all showed the same AP (AP1). In addition, XCGY1 to XCGY4 collected from the same area were divided into two different groups, which indicated that Xcc strains were also mixed in the same regions of China. Interestingly, based on the results of the cluster analysis of all strains, the predominant worldwide races 1 and 4 were not classified into AP1/DNA I.
The method of rep-PCR is a kind of fingerprint analysis with different genomes. It is used to amplify the highly conserved repetitive sequences that are widely distributed in the bacterial genome to reveal the differences among strain genomes (Versalovic et al., 1991). A study showed that rep-PCR presented potential to determine the diversity within Xcc similar to that of other techniques like AFLP. At present, REP, ERIC, and BOX sequences in rep-PCR are frequently used in polymorphism analyses of bacteria (Versalovic et al., 1991, 1994). These repetitive sequences, which are distributed at various sites in the bacterial genome and separated by various distances, are different at the species level. In a recent study, the rep-PCR fingerprint profiles showed a high level of diversity of the 75 Xcc strains collected from India (Rathaur et al., 2015). In our study, these strains were clustered with a 90% similarity coefficient into 14 groups. Cluster analysis based on rep-PCR showed that there is a diversity of Xcc strains in different agro-climatic regions in China.
From the pathogenicity tests, ten representative strains XCNM1, XCTY, XCGY3, XCJY, XCNJ, XCGZ1, XCWL, XCYL, XCCW, and XCDX1 collected from 10 distinct provinces, were virulent to three hosts and had no significant correlation with the regional distribution. These Xcc strains in the same clade demonstrated similar virulence to different hosts, while the differentiation of pathogenicity between strains in separate clades was significant for host WJ0006. For instance, race 1 and XCTY belonging to Clade 2 presented high-pathogenicity to three brassica hosts, while strains XCJY, XCNM1, XCGZ1, XCWL, XCYL, XCCW, XCDX1 in Clade 1 displayed low-pathogenicity to inbred line WJ0006 and high-pathogenicity to inbred line XGBH and 87-534. The diversity of pathogenicity of the Xcc strains to the host and the genetic diversity of resistance to black rot in different materials illustrated that the resistance response in brassica crops is related to both the genotype of the hosts and the virulence of the Xcc, as a result of the interaction between the host and the pathogen.
At present, black rot disease is widely distributed in China, but little is known about the pathogen genetic diversity and population structure. In this study, we collected Xcc strains from all over China, and compared them with 7 representative Xcc strains derived from America, Europe, and Australia. An interesting result was obtained using two genotyping methods, which showed that almost 80% of Xcc strains collected from China were divided into the same group, specifically, up to 31 strains with genotypes belonging to both AP1 and DNA I were distinguished from 7 race strains originated from abroad regions. The data analysis showed that the two genotyping methods had a strong correlation, and they could be used to distinguish Xcc strains. The results of virulence tests indicated that Xcc strains grouped in the same clade are similarly virulent, and the pathogenicity varied among clades. The combined findings suggest that AP1 or DNA I (including 31 common strains) is the predominant Xcc group causing cabbage black rot in China. Moreover, The genetic and pathogenicity tests showed that only a few Xcc strains showed high similarity with known races, and most of the strains were divided into a separate group from races 1-6 and 9, suggesting that the Xcc population structure might be greatly affected by geographic conditions.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print