Isolation and Genomic Characterization of the T4-Like Bacteriophage PM2 Infecting Pectobacterium carotovorum subsp. carotovorum
Article information
Abstract
In order to control Pectobacterium carotovorum subsp. carotovorum, a novel virulent bacteriophage PM2 was isolated. Bacteriophage PM2 can infect 48% of P. carotovorum subsp. carotovorum and 78% of P. carotovorum subsp. brasilliensis but none of atrosepticum, betavasculorum, odoriferum and wasabiae isolates had been infected with PM2. PM2 phage belongs to the family Myoviridae, and contains a large head and contractile tail. It has a 170,286 base pair genome that encodes 291 open reading frames (ORFs) and 12 tRNAs. Most ORFs in bacteriophage PM2 share a high level of homology with T4-like phages including IME08, RB69, and JS98. Phylogenetic analysis based on the amino acid sequence of terminase large subunits confirmed that PM2 is classified as a T4-like phage. It contains no integrase- or no repressor-coding genes related to the lysogenic cycle, and lifestyle prediction using PHACT software suggested that PM2 is a virulent bacteriophage.
Pectobacterium carotovorum subsp. carotovorum (formerly known as Erwinia carotovora subsp. carotovora) is a Gram-negative plant pathogenic bacteria that causes severe soft rot diseases in various crops, resulting in serious economic losses (Lee et al., 2013). Although several methods are applied to control bacterial pathogens, bacteriophages are considered to be one of the most effective in terms of target specificity, economic feasibility, and safety (Frampton et al., 2012). Phages are very specific to its target bacteria, so they do not harm the useful bacteria lived in environment or animal body. When well-purified phages are used, side effects were rarely appeared for all types of administration. Bacteriophages can multiply when their host bacteria exist, so bacteriophage can be prepared cost-effectively and the number of phage can increase at target point. Despite of many advantages of bacteriophage, phages are not widely used as biocontrol agents, even though bacteriophage application was recently reintroduced. Narrow host range and rapid appearance of resistant bacteria have been a big obstacle for bacteriophage to be a effective biological control agent (Loc-Carrillo and Abedon, 2011; Lu and Koeris, 2011).
Recently, the number of papers dealing with genome analysis of bacteriophage is increasing (Klumpp et al., 2012). Development of biotechnologies and collection of bio-information may help to overcome limitation of bacteriophage application by forming the basis of bacteriophage engineering. For example, engineered T7 bacteriophage to express an capsule degrading enzyme could take expanded host spectrum (Scholl et al., 2005), and modified reporter phages showed improved bacterial pathogen detecting ability and high sensitivity (Edgar et al., 2006; Kim et al., 2014). Comprehensive understanding of phage-host interaction and phage-resistant mechanism based on genomic information may suggest a key clue to lower phage-resistant bacteria appearance. In addition, bacteriophage genome analysis enables comparative genomics between bacteriophages or phage and its host bacteria to investigate evolutionary diversity (Farlow et al., 2014; Yuan et al., 2014). Prediction of bacteria lysis associated genes such as endolysin, bacterial cell wall degrading enzyme produced from bacteriophage, raises a change of antibiotic derivative development. However, though the availability of full genome sequence data has increased the potential of bacteriophages to control pathogens, only a small number of bacteriophages that target P. carotovorum subsp. carotovorum have been studied (Lee et al., 2012a; Lee et al., 2012b; Lim et al., 2014). Here, we report the isolation of new Myoviridae bacteriophage PM2, and characterization of the genome.
Bacteriophage PM2 was isolated from soil samples which were collected from Chinese cabbage fields in Pyeongchang, South Korea using P. carotovorum subsp. carotovorum as host bacteria. The bacteriophage was isolated as described previously, with minor modifications (Lim et al., 2013). Briefly, soil sample (5 g) was added with TSB broth (10 ml) and MgSO4·7H2O (final 10 mM). After weak homogenization in a BagMixer 400 blender (Interscience Laboratory), one milliliter overnight culture of P. carotovorum subsp. carotovorum was mixed, and initial propagation was performed for 24 hr. Isolated bacteriophage was confirmed by plaque assay and purified by picking plaques and elution in SM buffer repeated five times (Kropinski et al., 2009). Genomic DNA was isolated from the bacteriophage PM2 using phenol–chloroform extraction (Wilcox et al., 1996). The extracted DNA was sequenced using a Genome Sequencer FLX (Roche, Mannheim, Germany), and the qualified reads were assembled into a complete genome sequence using Newbler ver. 2.3 (Macrogen Inc., Seoul, Korea). Open reading frames (ORFs) were predicted using Glimmer 3 (Delcher et al., 2007), GeneMarkS (Besemer et al., 2001), and FgenesB software (Softberry Inc., Mount Kisco, NY). The function of the predicted ORFs was then assessed using BLASTP (Altschul et al., 1990) and Pfam domain prediction. The tRNA coding region was predicted using tRNAscan-SE software (Schattner et al., 2005). Phylogenetic analyses of the terminase large subunit of different bacteriophages was performed using MEGA5 based on the neighbor-joining method (Kumar et al., 2008). The lifestyle (temperate or virulent) of PM2 was predicted using PHACTS software (McNair et al., 2012).
Transmission electron microscopy images of Phage PM2 revealed that it has a head ~90 nm in diameter and a contractile tail ~90 nm in length and it was classified into the Myoviridae family in the order Caudovirales (Fig. 1). This phage has a bigger head and a shorter tail than previously reported P. carotovorum subsp. carotovorum phage PM1 that has a head ~55 nm in diameter and a contractile tail ~120 nm in length (Lim et al., 2014).
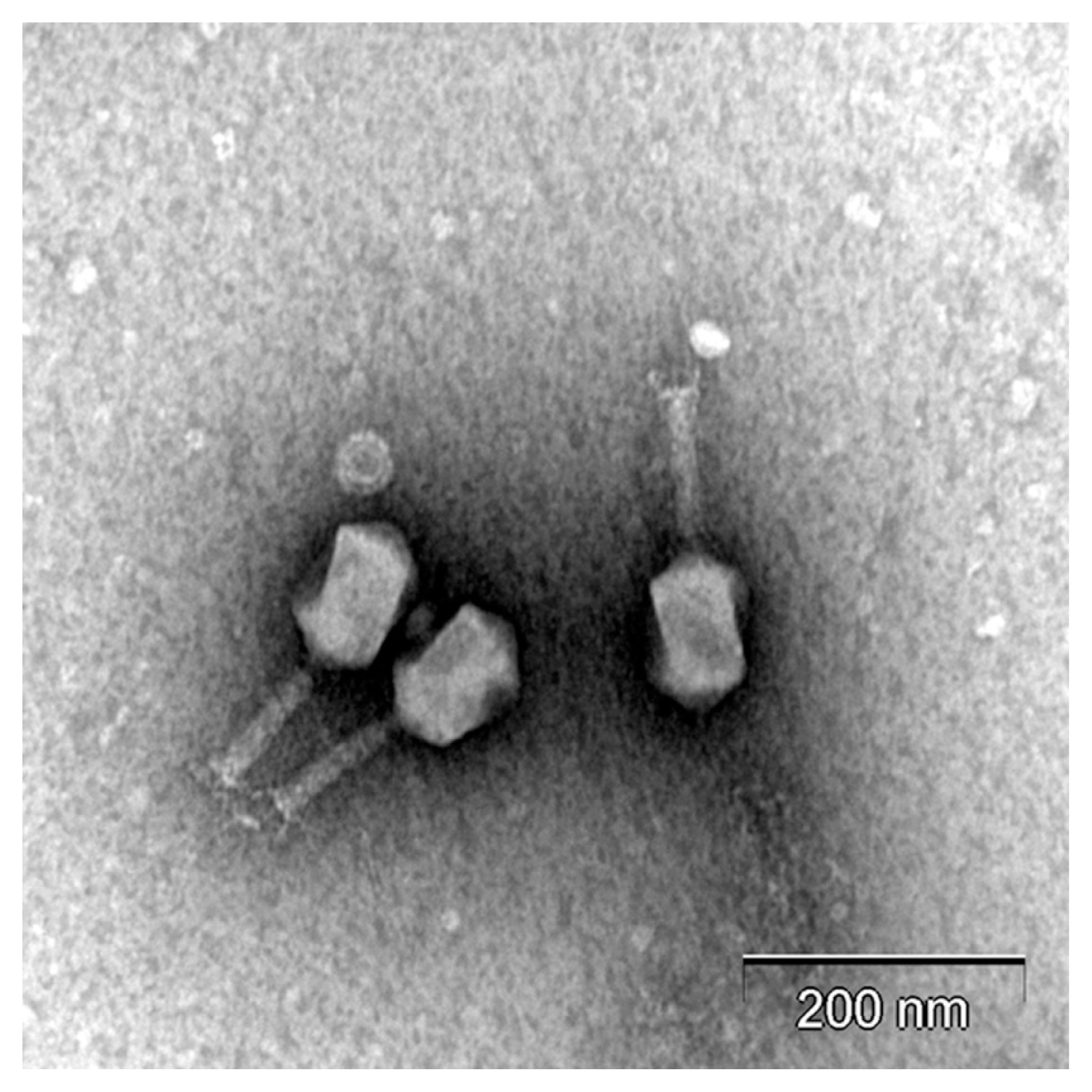
The morphology of bacteriophage PM2. The phages were visualized by staining with 2% aqueous uranyl acetate (pH 4.0) and examined with a LEO 912AB transmission electron microscope (Carl Zeiss, Wetzlar, Germany). Scale bars = 200 nm.
The bacteriophage PM2 showed relatively narrow host ranges. It was able to infect 26 P. carotovorum subsp. carotovorum among 54 tested isolates that collected nationwide (Table 1). There were no relations between the collected area or host plants of bacterial strains and the susceptibility to Phage PM2. Though it was able to infect only 48% of tested carotovorum subspecies, it was able to infect 78% of brasilliensis strains but could not infect any of tested odoriferum, atrosepticum, betavasculorum and wasabiae isolates. This suggests that subspecies brasilliensis is very close to subspecies carotovorum.

The infectivity of bacteriophage PM2 against diverse Pectobacterium isolates
The PM2 genome consists of 170,286 base pairs (bp) that encode 291 ORFs and 12 tRNAs (GenBank accession number: KF835987). Average G+C content of the PM2 genome is 34.8%, and it is quite low value compared to host bacteria, P. carotovorum (52%). One hundred seventy-eight ORFs (61%) were annotated as hypothetical protein-encoding genes, and the functions of 113 gene products (39%) were predicted (Fig. 2 and Table S1). PM2 exhibited a high level of homology with T4-like enterobacteria phages including IME08, RB69, and JS98 (Fig. 3) (Chibani-Chennoufi et al., 2004; Jiang et al., 2011; Nolan et al., 2006). In addition, phylogenetic analyses of terminase large subunits (Fig. 4) and major capsid proteins (data not shown) suggested that PM2 phage could be classified as a T4-like bacteriophage (Casjens and Gilcrease, 2009). Though, bacteriophage PM2 is the secondly reported Myoviridae phage targeting P. carotovorum subsp. carotovorum following bacteriophage PM1 (Lim et al., 2014), there is not much homologies between the genome of two phages. The genome size of PM2 is three times bigger than that of PM1. Like the bacteriophage T4 and other T4-like phages, the PM2 genes could be categorized into several major groups based on their putative functions (Fig. 2). Many replication-related genes were predicted, including DNA topoisomerase II subunits (PM2_003 and PM2_285), DNA helicase, primase and related proteins (PM2_015; dda, PM2_031, PM2_038, and PM2_266), DNA polymerase and related accessory proteins (PM2_052, PM2_055, PM2_056, and PM2_057), and DNA ligase (PM2_220).
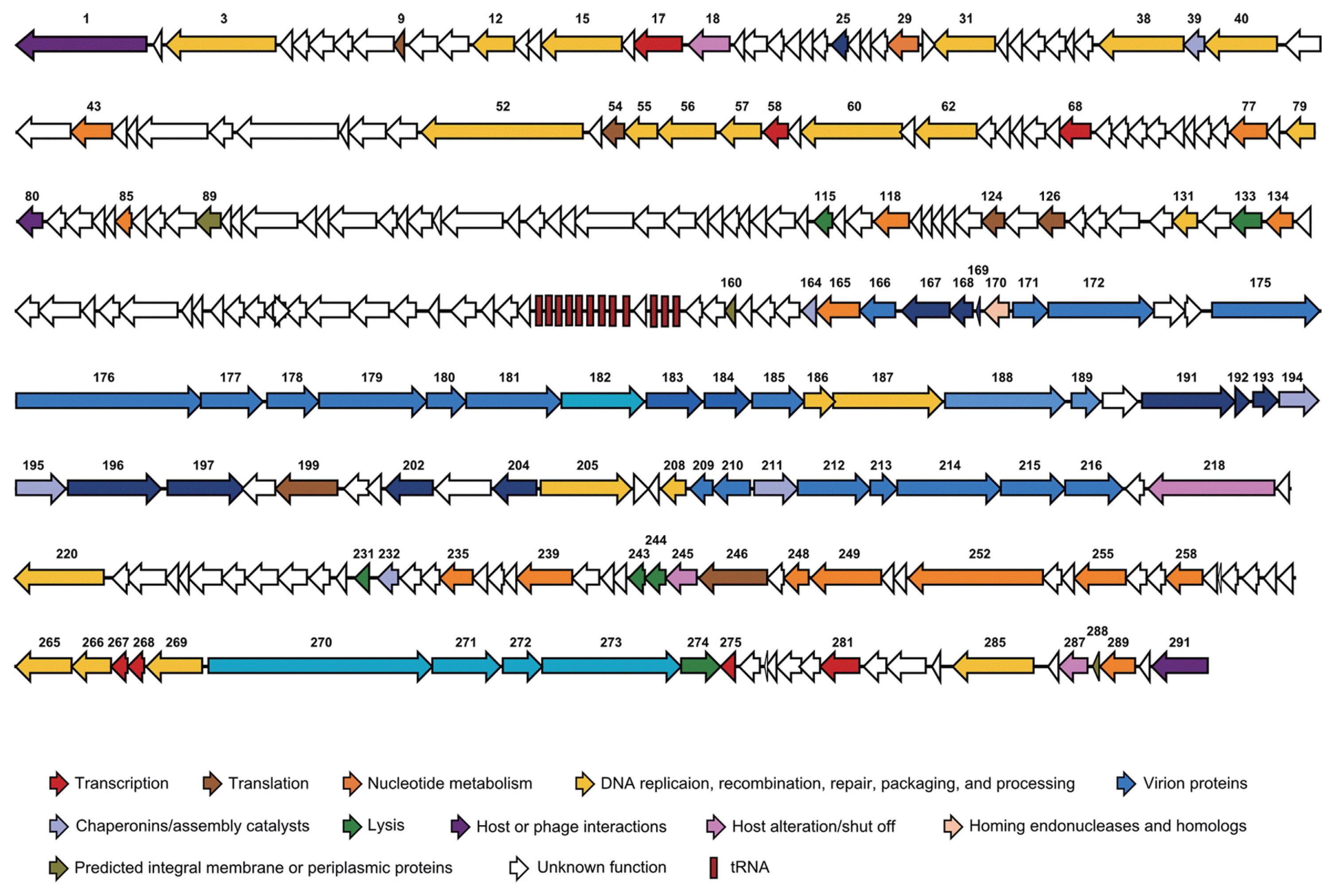
Genome map of bacteriophage PM2. Each ORF was categorized into functional groups based on information in available databases. Arrows indicate the position of genes related to transcription (red), translation (brown), nucleotide metabolism (orange), DNA replication (yellow), virion structure (dark blue, head; medium blue, neck; light blue, tail; pale blue, tail fiber), chaperonins (lavender), lysis (green), host or phage interactions (purple), host alteration/shut off (pink), homing endonucleases (peach), and membrane proteins (olive). Hypothetical proteins are colored white. The red rectangle indicates the tRNA-coding region.

Alignment of genome maps of phages PM2, IME08, and T4. Arrows indicate genes related to transcription (red), translation (brown), nucleotide metabolism (orange), DNA replication (yellow), virion structure (dark blue; head, medium blue; neck, light blue; tail, pale blue; tail fiber), chaperonins (lavender), lysis (green), host or phage interactions (purple), host alteration/shut off (pink), homing endonucleases (peach), and membrane protein (olive). Hypothetical proteins are colored white. DNA sequence similarities between the two genomes are indicated by gray shading.
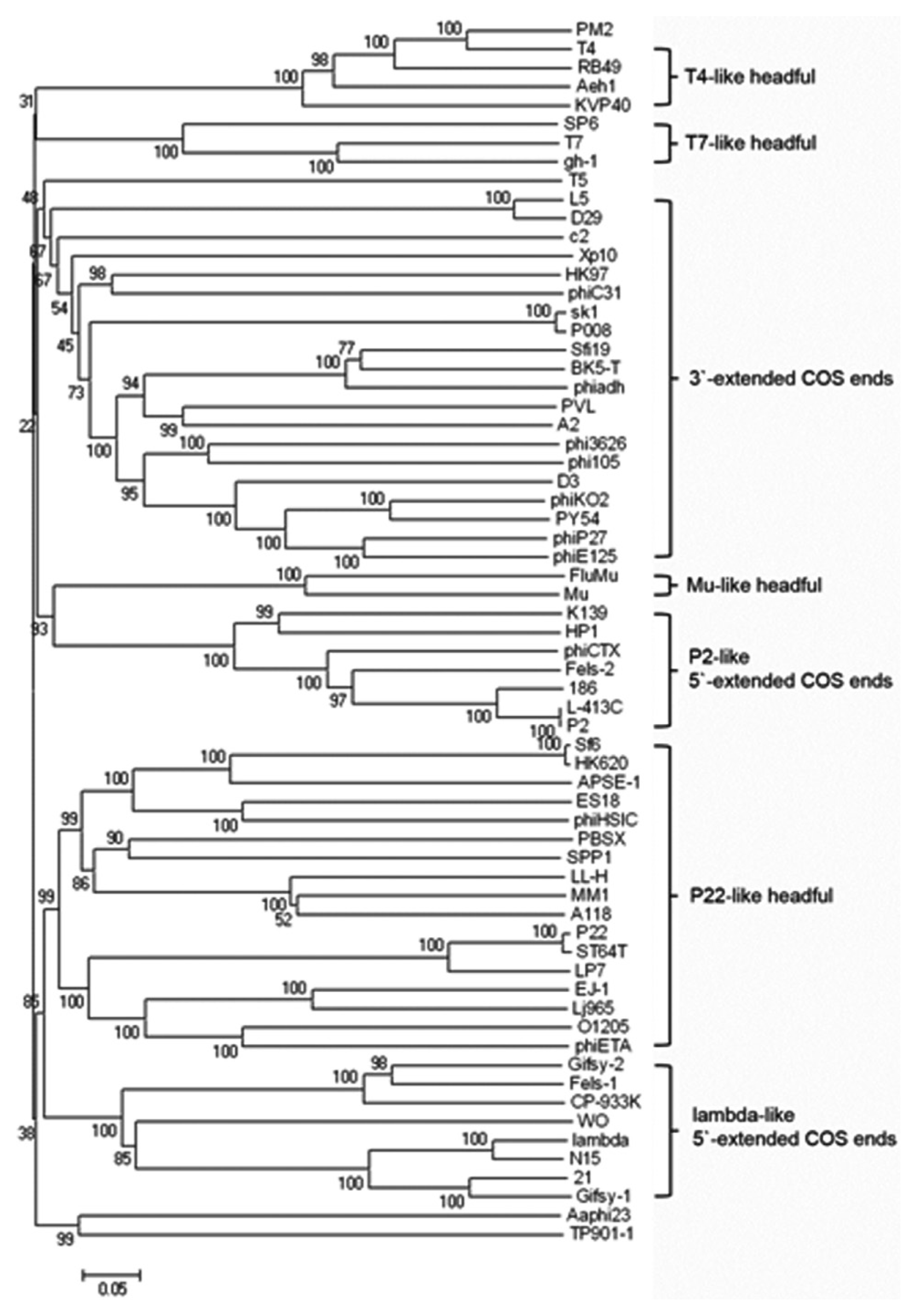
Phylogenetic analysis based on the alignment of the terminase large subunits amino acid sequence of phage PM2 and other phages. The sequences were compared using ClustalW, and the neighbor-joining phylogenetic tree was generated by P distance values using MEGA5.
Bacteriophage PM2 was predicted to contain several genes encoding proteins that are related to host alteration/shutoff. PM2_043 was annotated to encode thymidylate synthase, which is needed to convert cytosine into hydroxymethylcytosine (HMC) to evade host restriction endonucleases. HMC residues are then commonly modified further by glucosyltransferase in T4 phages; however, PM2 was not predicted to contain a gene encoding a glucosyltransferase. The product of PM2_245 was predicted to encode the transcription inhibitor Alc, which binds to RNA polymerase and DNA to terminate transcription from C-containing (host), but not HMC-containing (phage), DNA. Two genes were homologous to the anti-sigma factors (PM2_017; srd, PM2_275; asiA) of the T4 phage. AsiA inhibits the interaction between the host σ70 and the −35 region of the promoter by binding to the sigma factor. T4 Srd resembles a segment of σ70 and σ38, and shares interaction sites for the host sigma factor with RNA polymerase (Miller et al., 2003). Genes annotated as endonucleases, which are assumed to be responsible for host DNA degradation, were also identified (PM2_060, PM2_062, PM2_248; denA, PM2_289; denB).
Many proteins were also predicted to be involved in PM2 phage structure. The putative small outer capsid protein (PM2_025; soc), head outer capsid protein (PM2_202; hoc), and major head protein (PM2_196) are needed for head structure. For scaffold assembly, several prohead core proteins (PM2_192, PM2_193, PM2_194, and PM2_195) and an inhibitor of prohead protease (PM2_204; inh) were annotated. Four proteins (PM2_183, PM2_184, PM2_168, and PM2_169) were predicted to play roles in head completion. More than 20 different proteins were identified that were related to tail structure, including the tail tube and tail sheath monomer (PM2_189 and PM2_188, respectively), base plate hub or wedge subunits, and their connector and stabilizer. Most structural proteins were relatively conserved in T4 and IME08, whereas the tail fiber proteins (PM2_182, PM2_270, PM2_271, PM2_272, and PM2_273) exhibited low homology (Fig. 3). The tail fiber is the most important factor for phage–bacteria specificity. Therefore, the low similarity between the tail fibers of Pectobacterium phage PM2 and enterobacteria phages T4 and IME08 corresponds to their different host specificities.
Holin and endolysin, which play roles in host lysis, were predicted (PM2_274 and PM2_133, respectively). When holin makes pore bacterial cell membrane, endolysin can reach to its target, peptidoglycan. One of covalent bond composed of peptidoglycan is break down by endolysin, and finally cell was lysed (Ziedaite et al., 2005). In addition, Rz/Rz-1 like proteins, which are considered to be accessory proteins needed for the lysis of Gram-negative hosts, were also annotated (PM2_244 and PM2_243) (Summer et al., 2007). In lambda bacteriophage, the Rz is an integral inner membrane protein and Rz-1 is an outer membrane lipoprotein. Two proteins provide a physical connection between the inner and outer membrane by binding each other, and the complex span the periplasm. Under the certain condition, high concentration of divalent cations, Rz/Rz-1 like proteins are necessary for full lysis (Summer et al., 2007). PHACTS software predicted that PM2 phage is classified in the lytic lifestyle, similar to other T4-like phages (data not shown). Consistent with this prediction, no lysogenic cycle-related genes, such as an integrase or repressor, were identified. In addition, no toxin-related genes were observed.
In this paper we described the genetic structure of bacteriophage PM2 related with putative functions. These data comprise deep analysis about genes categorized into three groups; host alteration/shutoff, structural genes, and lysis related genes. Since endolysin lyses bacterial cell wall, this can be used as an effective antimicrobial against certain pathogen. Numerous publication has described how these endolysins can be used as an effective antimicrobial (Schmelcher et al., 2012). Endolysin engineering based on genomic data has opened a range of new applications from plant disease control to food safety. This paper provides information about bacteriophage host specificity with genes involved in the host alteration/shutoff. These genes may functions in the limitation of host ranges. Accumulations of many these kind of data will provide the clue to solve the problems of host specificity of bacteriophage as an effective biological control agents.
Acknowledgments
This work was supported by the Rural Development Administration (RDA) fund PJ008615. D. H. L. and J.–A L. were supported by the postdoctoral fellowship program of the National Academy of Agricultural Science (NAAS), RDA, Republic of Korea.