Potato virus Y (PVY; genus Potyvirus, family Potyviridae) is one of the most widespread and important viruses infecting agriculturally important plants belonging to the family Solanaceae including potato, tobacco, and pepper (De Bokx and Huttinga, 1981; Shukla et al., 1994). PVY is transmitted by at least 40 aphid species (family Aphididae) in a nonpersistent manner (Edwardson and Christie, 1997). Vertical transmission is also a serious problem for commercial tuber seed production in potato (Blanc et al., 1997). The monopartite genome is composed of a single-stranded, positive-sense RNA molecule of about 10,000 nucleotides long containing a single open reading frame (ORF) (Lorenzen et al., 2006). The encoded large polyprotein is processed by three virus-encoded proteases to produce at least 10 mature functional proteins (Urcuqui-Inchima et al., 2001).
Several distinct strains of PVY including tobacco veinal necrosis PVYN (N strain) and tuber necrosis strain PVYNTN, which induces potato tuber necrotic ringspot disease (PTNRD), have been recognized (Tian et al., 2011). Isolates of the N strain are not harmful only to tobacco but are also problematic in potato, where they overcome Ny and Nc genes conferring hypersensitive resistance to PVY isolates belonging to the strain groups PVYO and PVYC, respectively (Singh et al., 2008). Despite attempts to limit their spread (Singh, 1992), PVYN isolates are now distributed all over the world and are of a matter of great concern to growers because of frequent emergence of new variants via mutation and/or recombination with other PVY isolates (Singh et al., 2008).
Studies of the molecular evolutionary history of viruses are very complex as they involve understanding variation caused by mutation, recombination, selection pressure and adaptation (Moury et al., 2004; Moury et al., 2002). These studies help to understand important aspects of viral biology such as changes in virulence and geographical ranges that leads to ‘emergence’ of new epidemics. Hence, information on this aspect of viruses is essential to design management and control strategies in order to hinder their spread.
Potato (Solanum tubersum L.) is an agriculturally and economically important crop in Iran that is grown over 190,000 hectares with an estimated production of 5.5 Million tons (FAO, 2013). PVY is one of the most damaging potato viruses in Iran and is responsible for the main yield losses (Pourrahim et al., 2007). Numerous studies have focused on etiology, pathogenesis, ecology, molecular biology and control of the virus (Hosseini et al., 2011; Pourrahim et al., 2007); however, there are still gaps in our knowledge in terms of genetic variability and population structure especially at full-length genome level. In the current study, we used different experimentally evolutionary approaches in order to understand the factors that influence PVY variability. By measuring the selective pressure exerted on the proteins encoded by the PVY genome, we found that mutations resulting in amino acid substitutions also contribute to PVY evolution. This analysis rarely done on plant viruses can also help protein function and structure studies as well as virus epidemiology.
Materials and Methods
Virus isolates
Forty five Iranian PVY isolates were selected from our previous surveys conducted in Iran (Pourrahim et al., 2007) including 14 new isolates that were collected during 2009 to 2013 (unpublished data). The new isolates were tested for the presence of PVY by TAS-ELISA using PVYN, PVYO/C and PVYO-specific monoclonal antibodies (Neogen, UK). Isolates were assigned a subscript of O, N, C or NTN depending on the serological data. All samples were homogenized in 0.1 M K-phosphate buffer (pH 7.4) and mechanically inoculated onto tobacco (Nicotiana tabacum cvs. Samsun or Xanthi).
RT-PCR, cloning and sequencing
Details of the isolates including original host plants and the year of isolation are shown in Supplementary Table S1. The full-length genomic sequences of four PVYN strain (IRNH1, IRNH172, IRND41, IRNZ33) were amplified by RT-PCR from the total RNA isolated from systemically infected N. tabacum cv. Xanthi leaves using an RNeasy Plant Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The RNAs were reverse transcribed and PCR amplified using high-fidelity PlatinumTM Pfu DNA polymerase (Invitrogen, Carlsbad, CA, USA). The 5′ terminus of the PVY genome was obtained using a 5′ RACE kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions. The resultant amplicons were purified using the QIAquick Gel Extraction Kit (QIAGEN, Hilden, Germany) and subjected to direct sequencing or cloned into EcoRV site of plasmid pZErO-2 (Invitrogen, Carlsbad, CA, USA), transferred into E. coli strain DH5α and plated on media containing 25 μg ml−1 kanamycin. Plasmids were purified using QIAprep Spin Miniprep Kit (QIAGEN, Hilden, Germany). PCR products or cloned fragments were sequenced by a primer walking approach in both directions using the BigDye Terminator version 3.1 Cycle Sequencing Ready Reaction Kit (Applied Biosystems) and an Applied Biosystems Genetic Analyser DNA Model 310. For sequencing of each PCR fragment different forward and reverse primers with 400 to 450 nt lengths apart were designed (data not shown) and sequences overlapping by at least 100 bp were obtained. Sequence data were edited and assembled using BIOEDIT version 5.0.9 (Hall, 1999).
Detection of recombination
The genomic sequences of PVYN isolates that were obtained in this study and those of 54 full-length sequences obtained from GenBank were used for recombination analyses (Table S1). Relationships of aligned polyprotein sequences were calculated separately using Neighbor-Joining (NJ) and Maximum Likelihood (ML) methods. Amino acid sequences were aligned using CLUSTAL X2 with TRANSALIGN for optimal alignment (Weiller, 1998). Recombination events, putative parental isolates of recombinants, and recombination break points were analyzed using several methods implemented in the RDP3 version 3.44b (Martin et al., 2010) with default configuration and Bonferroni corrected P-value cut-off of 0.05 and 0.01. Only those potential recombination events detected by at least three different methods with an associated P value of 1.0 × 10−6 were considered, and involving fragments sharing ≥97% sequence identity with their parental sequences were considered. All putative recombinants identified by the RDP3 program were rechecked using SISCAN version 2 (Gibbs et al., 2000).
Phylogenetic relationship and genetic variability
Phylogenetic relationship was inferred using the NJ, and ML methods implemented in the MEGA 5 (Tamura et al., 2011) and ML tree is shown (Fig. 3). We used Pepper mottle virus (PeMoV) (GenBank accession no. NC_001517) as an outgrop. Pairwise genetic distances were analyzed by the Kimura’s two-parameter method using MEGA v. 4.1 program (Tamura et al., 2007). DNASP version 4.10 (Rozas et al., 2003) was used to estimate haplotype diversity which was calculated based on the frequency and number of haplotypes in the population.
Analyses of selection pressures
The direction and strength of selection is measured by ω, the nonsynonymous to synonymous substitution rate ratio (dn/ds = ω), with ω < 1, = 1, and > 1 indicating purifying selection, neutral evolution, and positive selection, respectively. The non-synonymous to synonymous nucleotide substitution rate ratio (ω) was assessed using an ML codon-substitution model implemented in the CODEML program of the PAML4 package (Yang, 2007). Models M0, M1 and M7 do not allow for the existence of positively selected sites. M0 calculates a single ω ratio (between 0 and 1) averaged over all sites, M1a (nearly neutral) account for neutral evolution by estimating the proportion of conserved (ω = 0) and neutral (ω = 1) sites, and M7 (β) uses a discrete beta distribution between 0 and 1 to model different ω ratios between sites. Three models M2a (positive selection), M3 (discrete), and M8 (β plus ω) account for positive selection by using parameters that can estimate ω > 1 (Wong et al., 2004; Yang et al., 2000; Yang et al., 2005). The first step in identifying amino acid sites under positive selection is to test whether sites exist with ω > 1 by application of likelihood ratio test (LRTs) to compare nested models, therefore, three LRTs (M3 vs M0, M2a vs M1a and M8 vs M7) were used to assess the models’ fit to the data, as described by Wong et al. (2004). Where the LRTs suggested positive selection, the Bayesian methods (Bayes Empirical Bayes - BEB) approach (Yang et al., 2000) was used to identify amino acids subjected to positive selection. Sites having high posterior probabilities (P > 90%) of belonging to the site class with ω > 1 are good candidates for positively selected sites.
Results
Biological and molecular characterizations of the Iranian PVYN isolates
Four isolates out of fifteen positive samples identified by monoclonal antibody against PVYN were selected. All of the samples induced vein necrosis in N. tabacum cv. Xanthi. The genomes of selected Iranian isolates were 9703-9707 nucleotides long. For recombination analysis, the terminal primer sequences including 5’UTR (189 nucleotides) and 3’UTR (328 to 332 nucleotides) were omitted. All of the motifs reported for different potyvirus genes and encoded proteins were found. The regions encoding the P1 (190-1014 nucleotides), helper-component proteinase (HC-Pro) (1015-2409 nucleotides), P3 (2410-3504 nucleotides), 6 kDa 1 protein (6K1) (3505-3660 nucleotides), cylindrical inclusion protein (CI) (3661-5562 nucleotides), 6 kDa 2 protein (6K2) (5563-5718 nucleotides), genome-linked viral protein (VPg) (5719-6282 nucleotides), nuclear inclusion a-proteinase (NIa-Pro) (6283-7014 nucleotides), nuclear inclusion b (NIb) (7015-8571 nucleotides) and coat protein (CP) (8572-9376 nucleotides) were 825, 1395, 1095, 156, 1902, 156, 564, 732, 1557 and 801 nucleotides long, respectively. Furthermore, the region from 2919 to 3149 (231 nucleotides) was determined for pipo ORF. Genome-wide pairwise nucleotide diversity analysis of the complete genome revealed three regions representing: (0.0-11.9), (11.9-15.9) and (15.9-23.0) nucleotide diversity for N, O and C lineages, respectively. The lowest nucleotide diversity (0.0-4.0) was found for N-Europe followed by 4.0-7.9 for W (PVYN-Wi) and 7.9-11.9 for N-Europe & North America populations from N lineage. The highest nucleotide diversities were found for O and C lineages (Fig. 1).
Recombination analysis
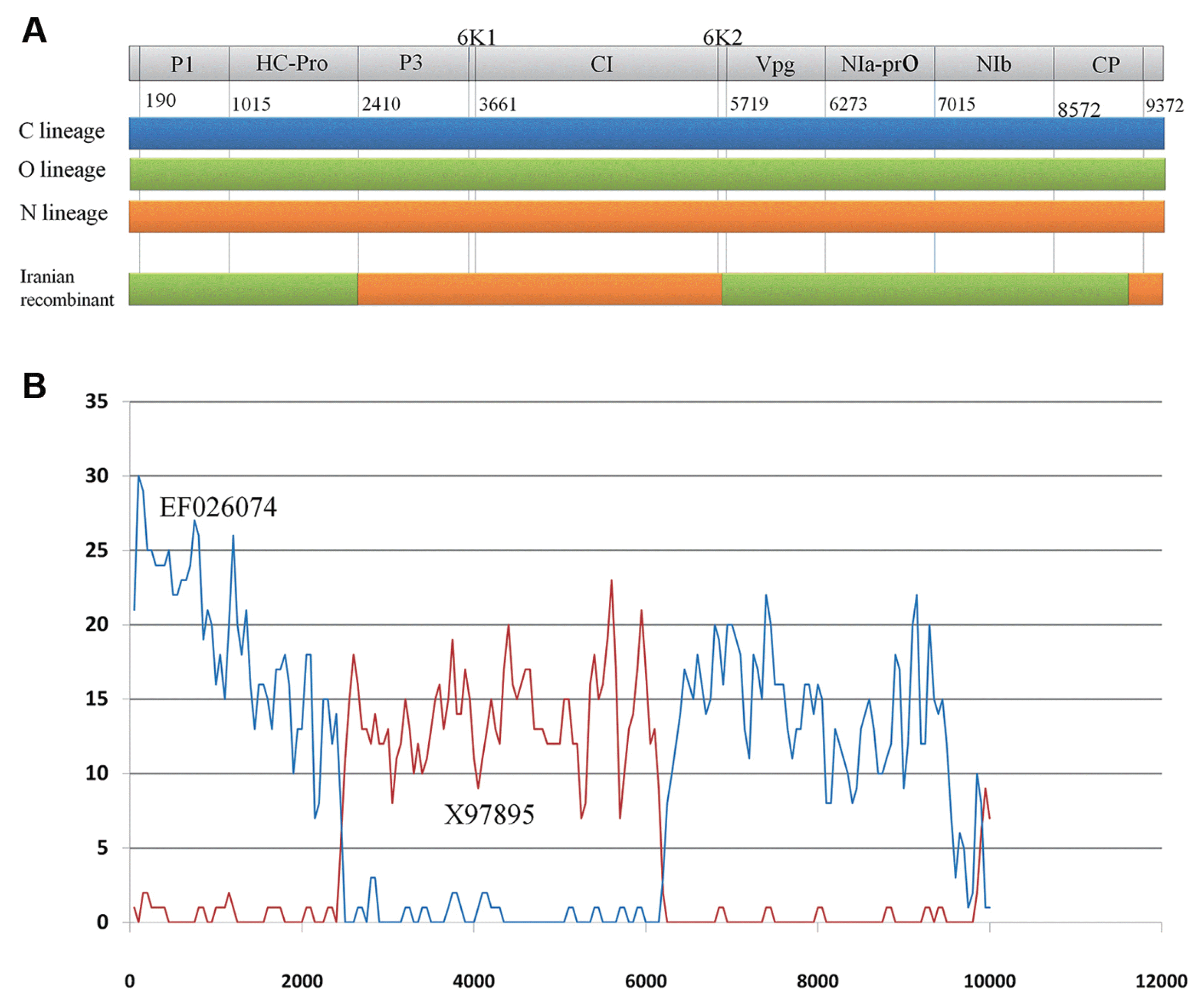
Different methods were used for recombination breakpoint prediction and provided evidence for clear recombination (Fig. 2, Table 1). Remarkably, following the adopted criteria (detectable by at least three different methods and recombined fragments with ≥ 97% nucleotide identity with parental sequences), recombination events observed across all four Iranian viral genomes analyzed. Recombination analysis demonstrated that Iranian PVYN had a typical European PVYNTN genome having three recombinant junctions, while PVYN and PVYO were identified as the parents. The recombination breakpoints were detected between P1 and HC-Pro (at nucleotide positions 190 and 2244), 6K2 and CP (at nucleotide positions 5581 and 8977), and in the CP (nucleotide positions 8977 and 9703) (Table 1). Every event identified by the RDP3 program was confirmed by SISCAN V2 with parental-like Oz (a Canadian isolate with accession no. EF026074) and a Switzerland isolate CH-605(NR) (accession no. X97895) from O and N lineages, respectively (Fig. 2A and B).
Phylogenetic relationships
Pairwise comparisons using CLUSTAL X2 were performed and after being degaped, there were a total of 9165 positions in the final dataset. Phylogenetic trees showed that PVY isolates fell into three main lineages where four Iranian PVY isolates (IRNH1, IRNH172, IRND41, IRNZ33) were clustered with European NTN strain (Fig. 3). The pairwise genetic diversity within population were 0.074±0.002, 0.035±0.001 and 0.120±0.003 for N, O and C lineages, respectively (Table 2). The highest pairwise diversity for C lineage may indicate the isolates in this lineage were more genetically diverse than N and O lineages. In addition, the between-population diversities (Table 3) were identified that were different to the within population diversity, indicating that there was genetic differentiation between these populations. The highest nucleotide diversities 0.229±0.004 were determined between C and N-Europe populations (Table 3).
Selection analyses
The mean ω ratio differed greatly among different cistrons. Using model M0, ω values were 0.267 (P1), 0.085 (HC-Pro), 0.153 (P3), 0.050 (CI), 0.078 (VPg), 0.087 (NIa-pro), 0.079 (NIb) and 0.165 (CP) (Table 4). Model M0, used to assess selection pressures [maximum likelihood (ML) framework of codon substitution], and yielded the value ≥ 0.267 over all codon sites in the 10 cistrons indicating purifying selection (Table 4). For P1 protein, heterogeneity of selection pressure, tested by using M3 vs M0 (LRT), revealed that M3 fitted the data significantly better than M0. Model M3 suggested that a large set of sites (56.2%) were evolving under strong purifying selection (ω = 0.062), fewer sites (31.6%) under weak purifying selection (ω = 0.441) and only 1.2% of sites under positive selection (ω = 1.131). Comparison of M8 with M7 showed that 32 sites (14.3%) were under positive selection (ω = 1.047) (Table 4). By using the BEB inference (Yang et al., 2005) 22 sites of P1 protein were identified under positive selection. Additionally the Naive Empirical Bayes (NEB) analysis inference (Yang et al., 2000) under M8 provided 10 more sites for P1 protein under positive selection (Table 4). For HC-Pro, M3 suggested that a large set of sites (69.5%) were evolving under strong purifying selection pressure (ω = 0.010), 28.6% were under weak purifying selection pressure (ω = 0.237) and only 1.7% of sites were under positive selection pressure (ω = 0.829). Using the NEB inference five sites of HC-Pro identified under positive selection pressure with models M3 and M8 but the LRT statistics for these sites were not significant. For P3, 53.1% of sites were evolving under strong negative selection pressure (ω = 0.006), 36.5% of sites under purifying selection pressure and 1.03% were under positive selection pressure. Using M3, M2a and M8, BEB predicted the 35 amino acids residue under positive selection (Table 4). For CI and VPg proteins the majority of sites were found under strong negative selection pressure with ω = 0.014 and ω = 0.020, respectively. However NEB and BEB analysis identified some sites under positive selection pressure with M3 and M8, respectively (Table 4). Although a few sites were found under positive selection pressure for NIa and NIb proteins using M8; however, the LRT statistics for these proteins were not significant.
Comparison of M8 with M7 for CP showed that M8 fitted the data better than M7 and 16 sites were under positive selection pressure. Seven sites (Table 4) identified by using the BEB inference with probabilities of 95 to 99% and high ω ratio (ω = 1.369); however, for nine sites the LRT statistics were not significant (Table 4). No positive sites were found for 6K1 and 6K2 proteins under different models. Comparison of P3 amino acid sites between PVY lineages showed that most sites under positive selection belonged to N lineage; however, some of the motifs including PSYNT (amino acids 160 to 164), KLSATWYSYRAKRSITRY (amino acids 190 to 207) and RGAQV (amino acids 230 to 234) were conserved among the three lineages (Table 3). Except PSYNT motif, the two other ones (amino acids 190 to 207 and amino acids 230 to 234) belong also to the overlapping PIPO protein.
Discussion
PVY is considered one of the most harmful potato viruses. Dependent upon strains, time of infection and cultivars involved the virus can cause serious yield losses reaching to 80% (De Bokx and Huttinga, 1981). Although PVY isolates belonging to PVYN strain usually cause mild or no obvious symptoms in potato foliage, yield losses may be increased by emergence of new recombinant strains such as PVYNTN which are considered more aggressive than those of PVYN inducing potato tuber necrotic ring spot disease (PTNRD) (Tian et al., 2011). Furthermore the PVYN-Wi differs in virulence from older PVY isolates; however, serologically it is related to PVYO isolates (Chrzanowska, 1991). Analysis of previously surveyed potato cultivation areas (Pourrahim et al., 2007) and newly collected isolates during 2009 to 2013 revealed an increase in incidence of PVYN (unpublished data) in some provinces located in the West and Central parts of Iran. Furthermore, recently Hosseini et al. (2011) showed an increase in the incidence of different strains of PVY including the recombinant isolates from the Southern parts of Iran.
In this srudy, we analyzed the full-length sequences of four N strain isolates from Western and Central parts of Iran (Table S1). The genomes of the Iranian isolates were 9703 to 9707 nucleotides long with the presence of all the reported potyviral motifs. The coding region (polyprotein) of Iranian isolates excluded stop codon consists of 9183 nucleotide. Recombination plays a major role in the evolution of viruses by generating genetic variation, reducing mutational load and producing new viruses (Froissart et al., 2005; Garcia-Arenal and Palukaitis, 2008). The Iranian recombinant PVYNTN isolates are more closely related to the European than North American NTN isolates (Fig. 2 and Fig. 3). Like most of the European isolates (Ogawa et al., 2008), four Iranian NTN isolates analyzed in this study contained three predictable recombination breakpoints between the P1 and HC-Pro, 6K2 and CP, and in the CP cistrons (Table 1) with parental-like isolates belonging to O and N lineages (Fig. 2A and B).
Most of seed potatoes in Iran originated from seed tubers imported from Europe and this suggests the possibility that the Iranian NTN strain originated potentially from an ancestral European PVY strains. Furthermore the spread of PVY mostly occurs from infected to healthy plants within the same potato field via insect vectors (Broadbent et al., 1956, Thresh, 1976). Therefore, occurrence of additional infections in the Iranian potato fields derived from the original infected seed tubers cannot be ruled out (Pourrahim et al., 2007).
Phylogenetic trees showed that four Iranian PVY isolates were clustered in N-Europe subpopulation from N lineage (Fig. 3). The within-population diversity was lower than the between population diversity suggesting the contribution of a recent expansion after a bottleneck, namely a ‘founder effect’ on diversification of PVY isolates (Table 2, Table 3). Furthermore, nucleotide diversity was higher in C lineage as compared to O and N lineages, which could be explained on the basis of genetic differentiation between three populations. Phylogenetic analyses showed that N-Europe population with closely related isolates appeared in N lineage, as supported by the lowest nucleotide diversity, low pairwise nucleotide diversity and high haplotype diversity (Table 2). A combination for high haplotype diversity and low genetic diversity, assessed by mitochondrial DNA markers, is taken as evidence of a recent population expansion after a genetic bottleneck and this also was found for Turnip mosaic virus (TuMV) (Tomitaka and Ohshima, 2006) and Cauliflower mosaic virus (CaMV) (Farzadfar et al., 2014). These results are also confirmed by the genome-wide pairwise diversity analysis of the PVY isolates (Fig. 1).
Using PAML analysis most of the sites under positive selection pressure were detected in N lineage compared to those of C and O lineages (Table 4) as previously indicated by Moury and Simon, (2011). This may show the potential of N lineage for better fitness during interactions with hosts, vectors or occupation of new niches as discussed by Karasev et al. (2011). Also, selective pressure on the newly generated recombinant genomes may be so high that only a few recombinant types could survive and succeed in genome propagation (Hu et al., 2009; Karasev et al., 2011).
Regarding the diversity of functions and interactions of different proteins (Moury and Simon, 2011), it is not surprising that evolutionary constraints vary between potyviral proteins. The largest and lowest dn/ds ratios were found for P1 and CI cistrons, respectively (Table 4). Furthermore PAML analysis showed different sites within P1, P3 and CP proteins were under positive selection pressure. Amongst all of the potyvirid proteins, P1 proteinase is the least conserved protein in sequence and the most variable in size (Adams et al., 2005). Mostly this non-conservative protein is thought to have a contribution in successful adaptation of the potyviruses into a wide range of host species (Rohožkoá and Navrátil, 2011; Valli et al., 2007). Also, P1 is a serine protease that self-cleaves at its C-terminus and acts as an accessory factor for genome amplification (Verchot and Carrington, 1995). Based on analysis and observations, Ramírez-Rodríguez et al. (2009) suggested that the determinants of Tobacco necrosis virus may be localized at the 3′ end of the P1 gene. Most of the amino acid sites under positive selection pressure were found in C-terminal of the P1 which may contribute in the ability of HC-Pro to suppress RNA silencing (Valli et al., 2006) to increase the pathogenicity of heterologous plant viruses in synergistic interactions (Pruss et al., 1997).
Using M3, M2a and M8, BEB predicted the 35 amino acids residue under positive selection pressure for P3 and interestingly most of thse sites overlapped P3N-PIPO protein (Table 4). Studies on P3N-PIPO have indicated that both P3 and P3N-PIPO are essential proteins for potyvirus infection with the potential for involvement in virulence of viruses to overcome resistance mediated by cyv1 and sbm-2 genotypes of pea (Choi et al., 2013). In addition, mutations that knock out expression of the PIPO protein in Turnip mosaic potyvirus but leave the polyprotein amino acid sequence unaltered are lethal to the virus (Chung et al., 2008; Vijayapalani et al., 2012). However, mutational analysis of the putative pipo of Soybean mosaic virus suggests disruption of PIPO protein impedes movement, but does not abolish replication in a single cell (Wen and Hajimorad, 2010).
The detailed evolutionary knowledge of eIF4E and VPg makes this coevolutionary pathosystem among the best studied in all of plant virology (Moury et al., 2004). It was shown the VPg of PVY to be under positive selection pressure (Table 4). Thus, mutations at the amino acid positions subjected to positive selection pressure in the VPg of PVY can be expected to play a role in PVY-plant interactions. Using infectious cDNA molecules from two PVY isolates differing in their virulence, Moury et al. (2004) showed that a single nucleotide change corresponding to an amino acid substitution (Arg119His) in the central part of the viral VPg was involved in virulence toward the pot-1 resistance. Also, positively selected codons of VPg are directly involved in overcoming resistance eIF4E alleles (Ayme et al., 2007; Charron et al., 2008; Moury et al., 2004).
In PAML analysis, comparison of M8 with M7 for CP showed that M8 fitted the data better than M7 and 16 sites were under positive selection pressure (Table 4). Most of the positive selection sites identified in N-terminal of CP were the same as those previously reported by Moury et al. (2002). The N-terminal part of CP is a notable example of the multifunctionality of potyviral proteins. This part of CP is predicted to be exposed on the virion surface and involves in binding ligands and aphid transmission (Atreya et al., 1995), cell-to-cell and long-distance movement (Dolja et al., 1994, Dolja et al., 1995; Rojas et al., 1997) and/or assembly (Hofius et al., 2007); hence, it can be a target for selection by both host plants and insect vectors.
The first experimental validations of predicted positively selected codon positions were measured for CP of PVY using the impact on virus fitness of nonsynonymous substitutions at codon positions 25 and 68. It was shown that these positive sites were involved in adaptive trade-offs (Moury and Simon, 2011). Also, deletion of 25 amino acids of the CP of Tobacco etch virus (TEV) (8 of these amino acids correspond to amino acid positions subjected to positive selection in the PVY genome) reduced the speed of cell to cell movement in tobacco and completely abolished systemic movement (Dolja et al., 1994). Although some an aspartic acid-alanine-glycine (DAG) amino acid triplet, to be highly conserved among potyviruses and essential in their transmission by aphids (Atreya et al., 1995) was under sever negative selection (Moury and Simon, 2011) as indicated in our results.
Our analysis reported in this paper, to the best of our knowledge, provides the first demonstration of population structure in PVY strain N in mid-Eurasia Iran using complete genome sequences that highlights the importance of recombination and selection pressure in the evolution of PVY. The accurate pictures of phylogenetic relationships and comparisons among distantly related virus genomes present an important insight into basic evolutionary mechanisms. Furthermore the appearance of new genetic types not only as a result of recombination, but also mutation, demonstrate a possible high risk that must be taken into account while considering planning for efficient control programs.


 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print