More than 75% of all crops are pollinated by assorted animals such as bees, butterflies, and bats (Lautenbach et al., 2012). Recently, due to climate change and usage of agricultural chemicals, flower-visiting insects are becoming insufficient (Quinet and Jacquemart, 2020; S├Īez et al., 2019). Decline the pollinator of economic crops is directly linked to the yield, resulting in significant economic losses (Gallai et al., 2009). Furthermore, the drawback of cultivars which do not form pollen and limitation of cultivation condition of orchards have be leading to the increase the artificial pollination (Nam et al., 2019). Those accumulating evidence shows that the precise process of pollination and the use of high-quality pollen can affect fruit and seed set and improve the quality and the quantity of crops (Samneg├źrd et al., 2019).
Each organ of a plant, including pollen, is surrounding different microbial composition (Hardoim et al., 2015). Epiphytic and endophytic microbial structures of plant are variously composed according to the cultivation condition and environment of the plant (Hirsch and Mauchline, 2012). Among plant-associated microbes, certain beneficial microbes are absorbed from the external into the endophyte to promote the growth of the plant (Orozco-Mosqueda et al., 2018). Pathogens also break the plant tissues in order to infect the plant (Wei et al., 2018). In the case of pollenassociated microbes, in addition to directly entering the endophytic microbial community and transferred to the other plants through the process of dispersion (Donati et al., 2018). If the pollen contaminated with plant pathogens can cause severe diseases to spread into whole orchards during the artificial pollination process. For examples, Erwinia amylovora, a fire blight pathogen, and Pseudomonas syringae, which causes bacterial canker disease in kiwifruit, can be spread by pollen (Cellini et al., 2019). Microbes in the pollen are known to be vertically transferred from the parent plant to the next generation, as well as horizontally transferred to their own or other objects in the orchard (Frank et al., 2017). Addition, the microbes in pollen are associated with the fitness of surrounding pollinators (McFrederick and Rehan, 2016). Even human allergies are known to be affected by the difference in the microbial structure of pollen (Obersteiner et al., 2016). Thus, for artificial pollination, pathogens contaminated pollen can lead to disorder in the host plant and cause a major epidemic in orchards as well as human health (Marques et al., 2013). Those finding demonstrated that importance preventing the initial inflow of these pathogens in pollens, as they can be spread to a wider area by myriad environment conditions, such as wind and pollinator. Therefore, in the process of importing and exporting of the pollens for artificial pollination, quarantine service is required to prevent the inflow of foreign pathogens that pose a threat to biosecurity. So far, the most of research on pollen microorganisms only focused on plant species or type of pollinators. Research has been conducted to diagnose the major high-risk pathogens through quarantine services (Liao et al., 2018). However, information on microbial composition that present in the pollen remains poorly understood. Especially, the presence of putative plant pathogens in pollens are insufficient. In this research, microbial community structure and potential plant pathogens in the imported fruits pollen for artificial pollination were investigated through metagenomic approaches. The objectives of this research were (1) providing basic information microbiota community structure, (2) comparing microbial composition differences among the pollens, and (3) investigating putative plant pathogens in commercially imported pollens.
To compare the microbiota communities of pollen samples, four kinds of fruit pollen imported in both year of 2018 and 2019 from China were collected. The fruit pollens were included with apple (Malus spp.), kiwifruit (Actinidia spp.), peach (Prunus spp.), and pear (Pyrus spp.). In case of apple, peach, and pear pollens, they were collected through Wellplus Co. (Daejeon, Korea), and kiwifruit pollen were collected through Jeju-Biotech Co. (Jeju, Korea).
In case of pollen DNA was extracted using FastDNA SPIN Kit following manufacturerŌĆÖs instruction. To extract pollen DNA with Nycodenz (Zhao and Kuwana, 2003) (Axis-shield Diganostic Ltd., Dundee, UK), bacteria cell extraction buffer (1 M Tris-HCl pH 7.5 [final concentration of 50 mM] 20 ┬Ąl, Triton X-100 [Sigma-Aldrich, St. Louis, MO, USA] 10 ml, mass up to 1 l with sterile water and 139.8 ┬Ąl of 14.3 M 2-mercaptoethanol [final concentration of 2 mM] was added per l just before use) and Nycodenz solution (8 g of Nycodenz was dissolved in a 10 ml of 50 mM Tris-HCl pH 7.5) were used. Peptide nucleic acid (PNA) clamping probe (Panagene, Daejeon, Korea) was used simultaneously for PNA blocking treatment (von Wintzingerode et al., 2000). PNA blocked-16S rRNA PCR reaction was performed with 2 ┬Ąl of DNA, 1 ┬Ąl of 27F forward primer (10 ┬ĄM), 1 ┬Ąl of 1492R reverse primer (10 ┬ĄM) (Supplementary Table 1), 12.5 ┬Ąl of Kapa HiFi HotStart ReadyMix (Roche, Basel, Swiss), 2.5 ┬Ąl of pPNA (7.5 ┬ĄM, 5ŌĆ▓-ggctcaaccctggacag-3ŌĆ▓), 2.5 ┬Ąl of mPNA (7.5 ┬ĄM, 5ŌĆ▓-ggcaagtgttcttcgga-3ŌĆ▓), and sterile water to a final volume of 25 ┬Ąl. PCR program included an initial denaturation at 98┬░C for 3 min, 24 cycles at 98┬░C for 10 s, 78┬░C for 10 s (annealing temperature of the PNA blocker), 55┬░C for 30 s (annealing temperature of the 16S rRNA primers), 72┬░C for 1 min, and a final extension at 72┬░C for 5 min. DNA quality was confirmed using a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). For determine of exclusion plastic DNA, sequenced raw data were compared for ratio of chloroplast originated from hostŌĆÖs plant cells. Ratio of comparison was visualized through bar graph (ggplot2 package in R).
DNA extraction using only by the DNA extraction kit, chloroplast originated from plant cells accounted for 89.2%, 91.1% in peach pollen. Likewise, in pear pollen, chloroplast was confirmed for a high ratio of 85.2% and 84.6%. In the case of DNA extracted by the Nycodenz method, the chloroplast in peach pollen was identified for 36.7% and 30.7%. In pear pollen, 46.8% and 70.4% was confirmed, which was less than a treatment using only the DNA extraction kit. In addition, bacterial DNA that amplified through PNA blocking were not confirmed to be classified as chloroplast DNA in peach pollen, and accounted for a low ratio of 0.2%, 0.7% in pear pollen (Supplementary Fig. 1). Pollen-associated bacterial DNA was able to efficiently exclude the plant DNA derived from the pollen by using Nycodenz with PNA blocking. The blocking protocol is considered that high-quality amplicon refining could focus only on microbes by reducing the rate of sequence derived from the plant cells, thereby enabling for more accurate identification of the microbial community structures. To our knowledge, the blocking approaches contribute that the protocol of extracting pollen-associated microbial DNA was secured that has not been clearly established before. Additionally, our results found that each fruit pollen has an independent bacterial community structure based on the fruit species. However, it was also confirmed that the bacteria community of each fruit pollen was composed differently depending on the collection year. These results show that bacterial compositions are affected by both plant species and environmental factor. These findings suggest that continued investigation of the pollen microbial community is required.
A total of 24 samples consisting of fruit pollens (n = 3 per fruit species) which imported over a two-year period were amplified to 16S rRNA region. Pollen DNA was extracted through Nycodenz method and PNA blocking was performed with refining amplicon. The amplicons were sequenced using Miseq of Illumina platform using Herculase II Fusion DNA Polymerase Nextera XT Index Kit V2 (Macrogen, Daejeon, Korea). V4 region was used to construct the library and 515F forward primer (5ŌĆ▓-gtgycagcmgccgcggtaa-3ŌĆ▓), 806R reverse primer (5ŌĆ▓-ggactacnvgggtwtctaat-3ŌĆ▓) were used in this process. To verify the quality of the sequenced raw data, error rates were measured through DADA2 package in R program. Calculated error values were expressed through error frequency plot using DADA2 package. To annotating confirmed base, the Silva rRNA database was used for bacteria. The operational taxonomic units (OTUs) were clustered by a machine learning, which was divisive amplicon denoising algorithm (DADA; https://benjjneb.github.io/dada2/index.html). Each sequence and quality score indicating error rate in fastq format was machine learned and the sequence were corrected according to the learning. If the corrected sequence were equal, they are clustered into an OTU. iNEXT (iNterpolation and EXTrapolation, version 2.0.19) package was used for arranging abundance vector. Additionally, the curves of each sample were positioned at a clip through gridExtra (version 2.3) package. For graphically display complex phylogenetic sequencing data, Phyloseq (version 1.30.0) package was used for visualizing alpha diversity plots. In addition, the microbiome (version 1.8.0) package which assist with the data set of Phyloseq package was used. Indexes of observed OTUs, Simpson, Shannon was used for measuring alpha diversity. Statistically significant differences were identified through the Wilcoxon rank-sum test (Mann-Whitney). The cutoff of P-value was set to 0.05. To represent beta diversity, vegan (version 2.5-6) package was used for the analysis of the ecological data, and graphic-related Grid (version 3.6.2), GridExtra package was used for coordinate formation and graphing. Non-metric multi-dimensional scaling (NMDS) plot was used to represent the beta diversity (Bray-curtis distance) and the cutoff of P-value was set to 0.05 for verify a significant difference in distance between coordinates. microbiomeSeq (version 0.1) package that combines data from OTUs, taxa assignment, phylogenetic tree under different conditions using Kruskal-Wallis test was used for visualizing the abundance bar graph. In addition, devtools (version 2.2.1) package was used to develop the function of R program. Local contribution to beta diversity values were measured to demonstrate an ecological uniqueness. All sequence data has been deposited into GenBank (accession no. SUB922682). The source code of R for data analyses is available on GitHub at https://github.com/ekfks0125/2020_Kim.git.
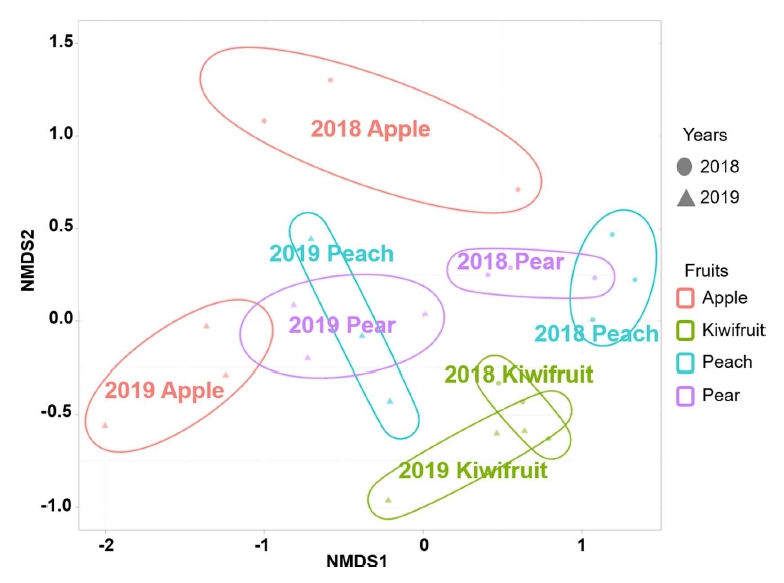
First, error frequency plot verified that each of the bases identified by the Illumina platform was classified as correct base (Supplementary Fig. 2). Through rarefaction curves, clustered OTUs were confirmed to maintain a constant number of OTUs when the read number was evaluated beyond the actual reading (Supplementary Fig. 3). Based on the assigned OTUs, NMDS plot represented the association of microbial structure of each fruit pollen. Even pollen from the same fruit species formed coordinates representing different microbial structures by the collected years (Supplementary Table 2). Regardless of the imported year, bacterial communities of apple-kiwifruit and apple-pear showed significant differences (Fig. 1). Richness, Shannon and Simpson indexes were used for analyzing alpha diversity (Supplementary Fig. 4). As results, bacterial structures were measured to have the lowest level of species abundance in the apple pollen. Shannon and Simpson indexes, which represent species richness and evenness, were measured higher in kiwifruit pollens with independent bacterial structures compared to the other fruits in 2019-year samples. In 2018-year, however, no significant difference was found for the evenness (Supplementary Fig. 4).
Taxonomic annotation at family level in 2018-year samples (Supplementary Fig. 5A), abundance bar graph confirmed that Enterobacteriaceae was the dominant bacteria in apple and kiwifruit pollens. In peach and pear pollens, Leuconostocaceae was showed as the highest abundance. in 2019-year samples (Supplementary Fig. 5B), peach and pear pollens-associated bacterial abundance pattern showed similar to the 2018-year sample. But in apple pollen, Burkholderiaceae was ranked at the highest rate. Additionally, kiwifruit pollen also showed different bacterial abundance pattern compare to the 2018-year sample. The results suggested that the pollen-associated bacterial community can be vary by fruit species and collection year. In the top 10 OTUs list, Enterobacteriaceae was measured at 12.4%, accounting for the highest composition in 2018-year apple. But in 2019-year apple, Burkholderiaceae were recorded 40.6% as the most abundance bacteria. Kiwifruit pollen presented that Moraxellaceae accounted for a large proportion in 2019-year sample. In both peaches and pears, Leuconostocaceae, equivalent to OTU1, accounted for more than 32% of the total in 2018-year samples, while in peach, OTU1 decreased to 12.6% in 2019, but in 2019-year pear, the OTU1 increased further to 39.6% (Table 1).
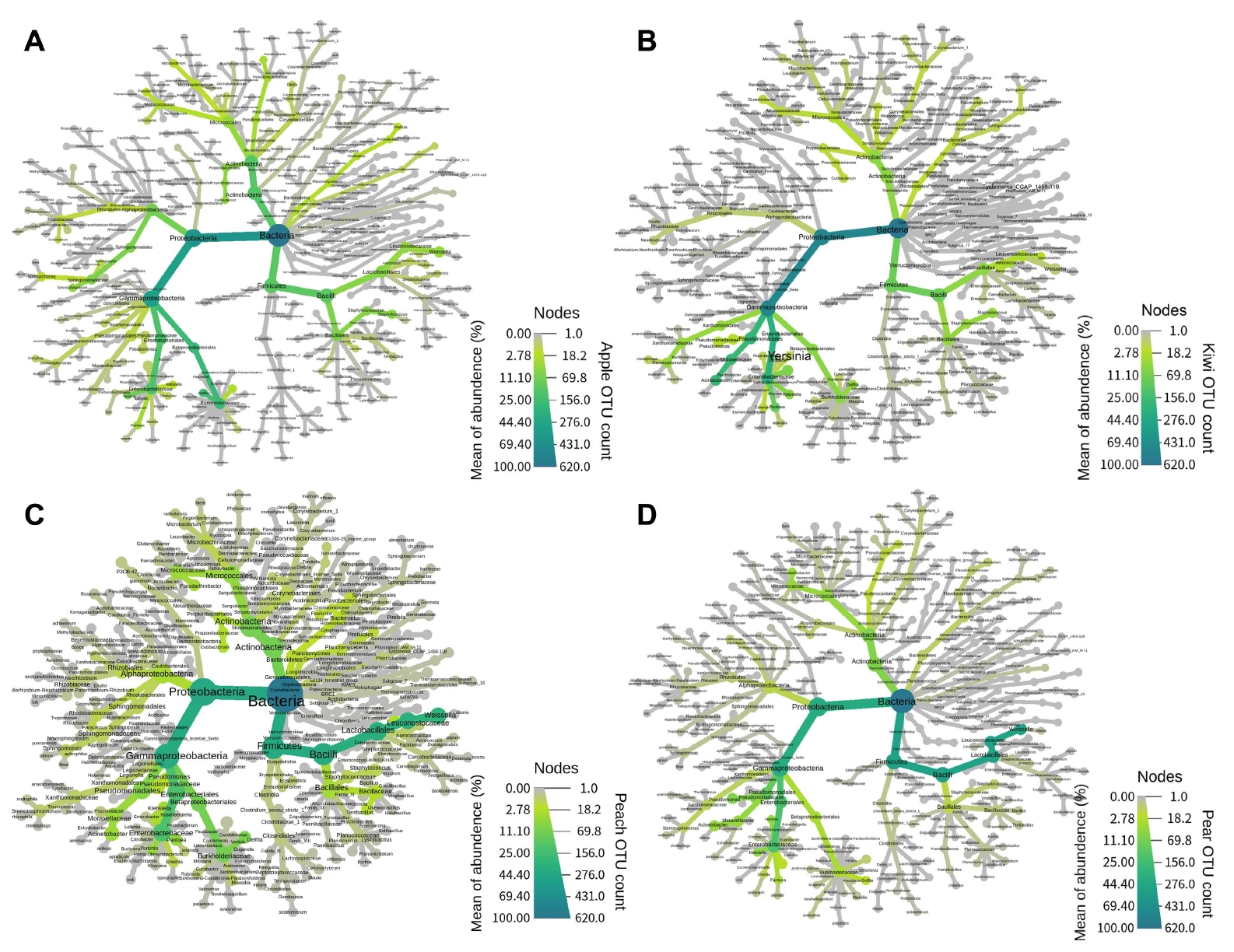
The metacoder (version 0.3.3) package was used to visualize the hierarchical structure of taxonomic classifications and identify the co-occurrence between the pollens. Predicted functional gene from sequenced pollen-associated bacteria was analyzed through Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2, version 2.1.0b). The predicted functional abundance was categorized by the KEGG BRITE and statistically significant difference between pollen samples was confirmed by Wald test of DESeq2 (version 1.12.3) package. The source code of R for data analyses is available on GitHub at https://github.com/ekfks0125/2020_Kim.git. As results of comparative analysis of pollen-associated bacteria species, peach pollen imported in 2018 were calculated a high degree of dominance value compared to other pollens (Supplementary Fig. 6). Dominance values of the kiwifruit pollen presented that the heat trees assigned to the Enterobacteriaceae were higher than other fruits. In the 2018-year pear sample had higher dominance of Actinobacteria than the other fruit pollens, but the 2019-year pear samples showed a lower dominance than all other fruits. Individual analysis of the metacoder by pollen species, the apple pollen was analyzed that Delftia sp.ŌĆÖs node belonging to the Burkholderiaceae had the highest dominance value (Fig. 2A). In the case of kiwifruit, Yersinia sp. and Acinetobacter sp. of Gammaproteobacteria were found to have the higher level of abundance that other trees (Fig. 2B). In pear and peach pollens, which represented similar bacterial community structures, heat trees of Bacilli were measured with a high degree of dominance value (Fig. 2C and D).
In the top 10 OTUs, Moraxellaceae in kiwifruit pollen was dominant then other fruit pollens. This is suspected to exist in a number of Acinetobacter spp. in kiwifruit pollen, which is a uniquely dominant bacteria species as the flower specific microbes. In pear and peach pollens, given a large number of Leuconostocaceae, the upper taxon of Lactobacillus spp., which is known to be frequently found as the animal-associated microbes, it is considered that pears and peaches were visited by pollination animals in the sampling environment (Vannette, 2020). Dominant bacteria can affect the entire community composition, family of Enterobacteriaceae, which are commonly found in all fruit pollens, has the ability to dominate microbiome community (Cernava et al., 2019). Certain bacteria species belonging to this family, such as Pantoea spp. and Erwinia spp. need to be attention because of their destructive pathogenicity.
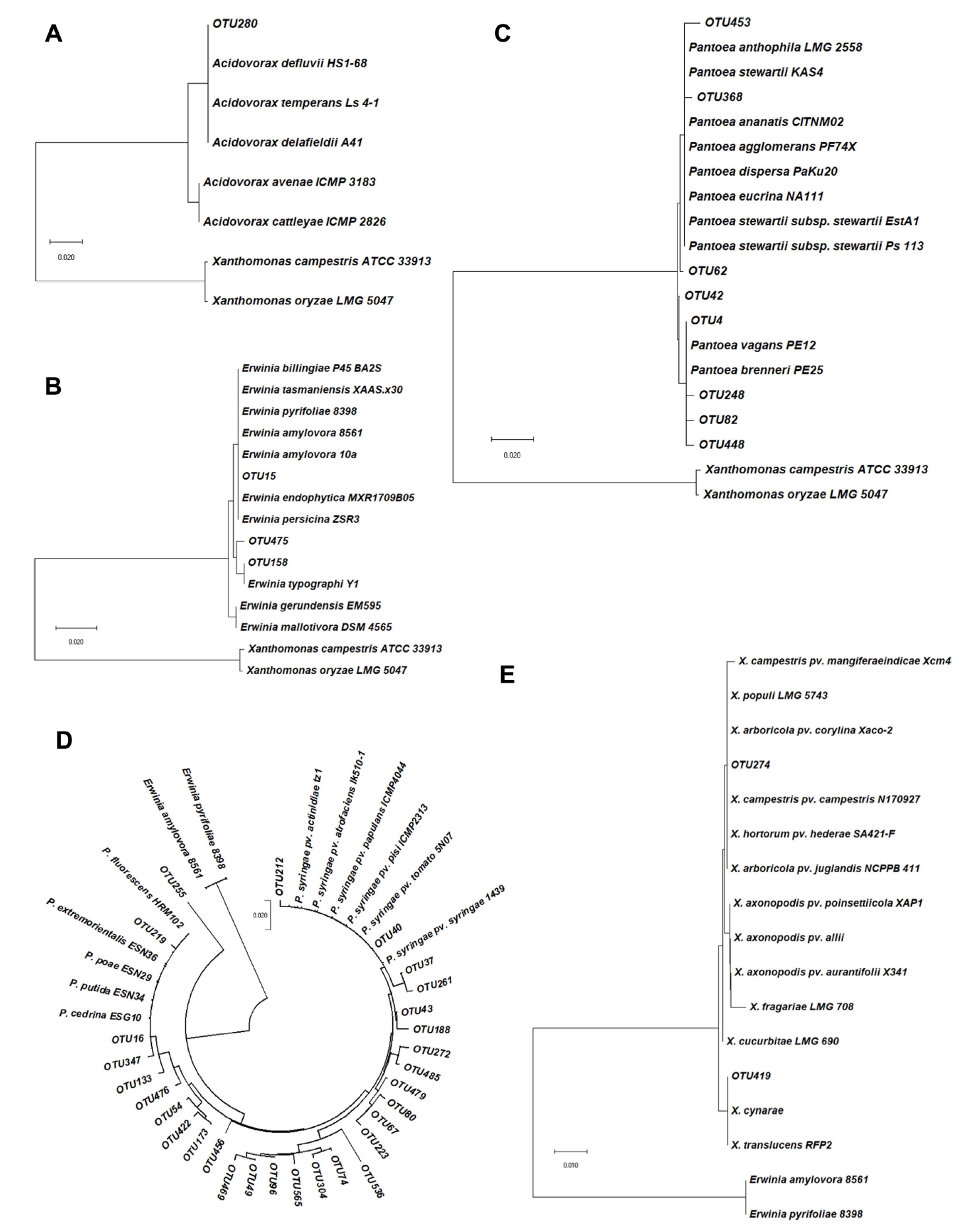
The list of prohibited pathogen species in quarantine service was registered by Animal and Plant Quarantine Agency (Korea). GenBank accession numbers for quarantine-protected species were descripted in Supplementary Table 3. Sequences were aligned through ClustalW (version 2.1) and phylogenetic trees were constructed by MEGA X (version 10.1) software with a Maximum Likelihood algorithm. Phylogenetic tree was analyzed through an alignment with bacterial species classified as the quarantine high-risk pathogens among the bacterial OTUs in the pollens. As results of the alignment with Acidovorax spp., although none of them were grouped with Acidovorax cattlevae, which classified to the quarantine prohibited pathogen. OTU280 in pear pollen was confirmed to be aligned with three kinds of Acidovorax spp. known as the plant pathogen (Fig. 3A). In the case of OTU15, which was detected in all pollens, phylogenetically belongs to same group to E. amylovora, high level of quarantine prohibited bacteria causing fire blight (Fig. 3B). For Pantoea spp., which contains numbers of plant pathogen, there was no OTUs that indicated a close association with a quarantine pathogen. However, OTU4, 82, 248, 448 in apple and pear pollens were analyzed to be closely related to pathogenic Pantoea spp. (Fig. 3C). For Pseudomonas spp., which contains quarantine management bacteria causing canker disease, OTU40 and OTU212 were paralleled with the putative plant pathogens (Fig. 3D). For Xanthomonas spp., OTU274 in kiwifruit pollen were grouped with Xanthomonas campestris pv. mangiferaeindicae known as causal pathogen of black spot disease in various plant and quarantine prohibited species. OTU419 in kiwifruit pollen were also grouped with the pathogenic Xanthomonas spp. (Fig. 3E).
Due to the characteristics of next generation sequencing using a relatively short region of 16S rRNA sequence, classification below the family level is not accurate. In the case of OTUs, which were confirmed to included high-risk pathogens, similarities to quarantine pathogens were identified using a phylogenetic tree. In addition to the commonly diagnosed target bacteria, many OTUs with high similarity to other plant pathogens have been detected. This evidence suggests that need for attention to the putative pathogens that have not been diagnosed so far. Through this study, our results provided basic information about the bacterial community in the pollens for artificial pollination in orchards. From a quarantine perspective, these findings suggest the need to block the influx of currently undiagnosed foreign pathogens. However, improvements will be needed to make information easier to use at quarantine sites by concisely visualizing metagenomic data through multiple analysis packages. The pathogen-oriented interpretation of the microbial structure of imported pollen is expected to contribute to the safe import and export of each country by establishing biological security.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement1
Supplement1 Print
Print