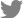Erwinia amylovora is the first Gram-negative bacteria found to cause disease in plants (Griffith et al., 2003; Hauben and Swings, 2005) and the causal agent of fire blight, a devastating disease characterized by tissue necrosis, of the Rosaceae family species, particularly apple (Malus domestica) and pear (Pyrus communis) (Griffith et al., 2003; Hauben and Swings, 2005; Johnson and Stockwell, 1998; van der Zwet et al., 1988). Fire blight was first reported in New York state (USA) in 1780, and then spread to other apple- and pear-producing regions around the world, including New Zealand, England, Europe, and the Middle East (Denning, 1794; Van der Zwet, 2002, 2006). Fire blight causes significant damage to the global apple and pear production annually. Production losses worth more than $40 million have been reported to date in USA, mainly in the states of Michigan, Washington, and Oregon (Longstroth, 2001; Norelli et al., 2003; Stockwell et al., 2002).
Initially, E. amylovora infects the tree trunk, shoots, leaves, flowers, and natural wounds through abiotic and biotic vectors, and then spreads through the vascular bundles, causing necrosis of the entire plant (Piqu├® et al., 2015; Thomson, 2000; Vanneste, 2000; Vrancken et al., 2013). The occurrence of necrotic lesions, bacterial ooze, tree canker, and blackened and curved shoot tips (ŌĆ£shepherdŌĆÖs crookŌĆØ) are typical symptoms of fire blight. Most of the initial infections occur through flowers, and the survival of pathogens in flowers and their relationships with the pre-existing microorganisms are considered important. However, because flowers are short-lived (5-10 days from flower opening to senescence), the number of microbial community studies on flowers is much less than that on roots and stems. Nevertheless, it has been established that flowers provide a unique environment for microbial communities (Cui et al., 2021; Shade et al., 2013).
Flowers attract pollinators and release various types of secretions such as nectar and pollen exudate to induce pollen germination (Cui et al., 2021; Shade et al., 2013; Vannette, 2020). These secretions are rich in sugars, amino acids, polysaccharides, and glycoproteins, thus serving as an excellent source of nutrients for many microorganisms (Borghi and Fernie, 2017; Vannette, 2020). A recent study showed that the diversity of the microbial community in the stigma of apple blossoms is relatively low, and a specific Pseudomonas lineage is dominant (Steven et al., 2018). During flowering, E. amylovora multiplies on the surface of the stigma and enters the plant (Spinelli et al., 2005). Therefore, the identification of abiotic and/or biotic factors that inhibit pathogen infection or development of fire blight is important.
It has been reported that frequent or long-term exposure of a non-target bacterial species to streptomycin can lead to the selection of a resistance gene, which can be transferred to the target pathogen such as E. amylovora (Chiou and Jones, 1993; Sundin and Bender, 1995). In addition, because of the outbreak and spread of E. amylovora strains resistant to streptomycin, an antibiotic mainly used to control fire blight, the use of plant control agents is nationally restricted (Chiou and Jones, 1993). Understanding the microbial community of fruit trees, as an alternative to the use of antibiotics, provides a solution for proper plant disease management and for the maintenance of immune homeostasis in fruit trees.
An important part of disease management is the suppression of primary fire blight pathogens through the use of microbial communities. However, little is known about how pathogen infection affects the microbial community of flowers. Previous studies on flower microbiome have generally been conducted using in vitro culture and 16S rRNA amplicon (V3-V4) sequencing methods (Pusey et al., 2009; Steven et al., 2018).
The advent of high-throughput sequencing technologies such as pyrosequencing, which analyzes bacterial communities through a non-cultivation method, offers a new perspective for microbial community analysis and significantly reduces the time and cost involved in profiling the diversity and abundance of microorganisms (Woese, 1987; Woese and Fox, 1977). Examining the taxonomic composition of bacterial communities by sequencing the hypervariable V3 and V4 regions of the 16S rRNA gene has been used to analyze microbial communities in various environments (Anderson et al., 2006; Barriuso et al., 2011; Schloss, 2010; Terlizzi et al., 2009). However, the most commonly used V3-V4 sequencing platform (Illumina MiSeq) is limited to analyze in the family or order level (Schloss, 2010).
Low-resolution classification limits the accuracy of ecological reasoning and the metabolic reconstitution and identification of appropriate bacterial strains. On the other hand, metagenomic shotgun sequencing, which analyzes the entire gene, provides high taxonomic and phylogenetic resolution but suffers from other disadvantages such as high cost (Teeling and Gl├Čckner, 2012; Wang and Jia, 2016) and a complex metagenomics-based community analysis (Teeling and Gl├Čckner, 2012). To perform higher resolution analyzes, a third-generation long-read sequencing technology developed by Pacific Biosciences RSII platform (PacBio, Macrogen Inc., Seoul, Korea) is being used for sequencing the 16S rRNA full-length gene (V1-V9). Recent studies show that 16S rRNA full-length gene sequencing produces higher taxonomic resolution and superior quality data than partial 16S rRNA gene sequencing (DŌĆÖAmore et al., 2016; Singer et al., 2016; Wagner et al., 2016).
In this study, we analyzed the microbial community of individual flowers collected from apple trees using PacBio sequencing. The microbial community of infected flowers was compared with that of uninfected flowers to understand the effect of E. amylovora on other microorganisms at the species level, with high resolution.
Three apple trees showing fire blight symptoms were selected from an orchard in Chungju, Chungcheongbuk-do, Korea, to collect diseased and healthy flowers. The temperature of the orchard at the time of sampling was 25 ┬▒ 5┬░C. The diseased apple blossoms showed fire blight symptoms on the sepals, pedicel, and receptacle. The healthy flower samples showed no symptoms of fire blight and were in close proximity to diseased flowers (within 30 cm). Each sample was analyzed by individual flower (100-200 mg/ flower), and in the case of diseased samples, it was confirmed that the overall degree of flower disease was discolored to black. The collected healthy and diseased flowers were suspended in sterile distilled water, and the suspension was centrifuged at 8,000 rpm for 10 min. The pellet was stored at ŌłÆ80┬░C until needed for microbial community analysis.
Microbial genomic DNA was extracted from diseased and healthy flower samples using the DNeasy Power Soil Kit (Qiagen, Norway). The PCR products were purified using the CleanPCR Kit (CleanNA, Waddinxveen, The Netherlands), and equimolar concentrations of the purified PCR products were pooled together. Non-target fragments were removed using the CleanPCR Kit, and the quality and size of the PCR products were evaluated using DNA 7500 chip in Bioanalyzer 2100 (Agilent, Palo Alto, CA, USA). Amplicons were manufactured using the PacBio platform, according to the manufacturerŌĆÖs instructions, and sequenced by Macrogen, Inc. (Seoul, Korea). The data generated were analyzed using the PacBio Single Molecule Real Time (SMRT) analysis portal (https://smrt-analysis.readthed-ocs.io/en/latest/SMRT-Analysis-Software-Installation-v2.3.0/), an easy-to-use web-based platform. The quality of sequence reads was assessed by FastQC, and a low-quality cutoff score was determined. Then, sequence reads were analyzed by QIIME2 (v 2020.2) (Bolyen et al., 2019) for quality control, diversity analysis, and taxonomic classification. The quality control functions of DADA2 (Callahan et al., 2016) were used to eliminate sequence noise, and to detect and eliminate chimeras. Community abundance was determined by measuring alpha diversity estimates, such as abundance-based coverage estimator (ACE) and operational taxonomic units (OTUs), and community uniformity estimates such as the Shannon index and inverse Simpson index. Phylogenetic tree was developed in QIIME2 to estimate beta diversity. Paired sample estimates (beta diversity) included the Bray-Curtis dissimilarity distance matrix and UniFrac.
Sequences were assigned to different species using the Silva 132 Reference taxonomy database (https://docs.qiime2.org/2019.1/data-resources/). DESeq2 was used to normalize the samples within the diseased and healthy group. Relative fold change (FC; P < 0.05) was used to identify taxa features expressed differently between diseased and healthy samples. Based on microbial community data, functional gene family abundance was predicted using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 (PICRUSt2) software (Douglas et al., 2020). The predicted functional profiles were obtained with the PICRUSt2 script, with default options (picrust2_pipeline.py), using bacterial community analysis and OTU tables of representative sequences. Next, the metabolic pathways from all domains of life were deduced from the predicted MetaCyc abundance using the ŌĆ£pathway_pipline.pyŌĆØ option. Analysis of variance (ANO-VA) of experimental datasets was performed using the R software (v3.6.3). The magnitude of the F-value (P < 0.05) determined the statistical significance of the treatment. The Shannon index, inverse Simpson index, and ACE metric were calculated using QIIME2. The distance between samples was calculated using the UniFrac algorithm. Values for sequence read number were followed by the Tukey post hoc test using R statistical software.
To evaluate the effect of fire blight on the microbial community of apple flowers, the 16S rRNA gene was sequenced in three replicates of diseased and healthy flowers. A total of 25,209 reads were categorized using the Protrimmer program. After the removal of chimeric and chloroplast genome sequences, a total of 20,949 sequences were obtained. Subsequently, the reads were clustered into amplicon sequence variants (ASVs), and those with > 99% sequence similarity were excluded from the analysis, resulting in 39 OTUs. Rarefaction curves were constructed for each individual sample showing the number of observed OTUs (Fig. 1A). Bacterial communities in flowers were much less diverse than those of rhizosphere bacteria. Rarefaction curves assessing OTU abundance per sample appeared close to saturation (Fig. 1A).
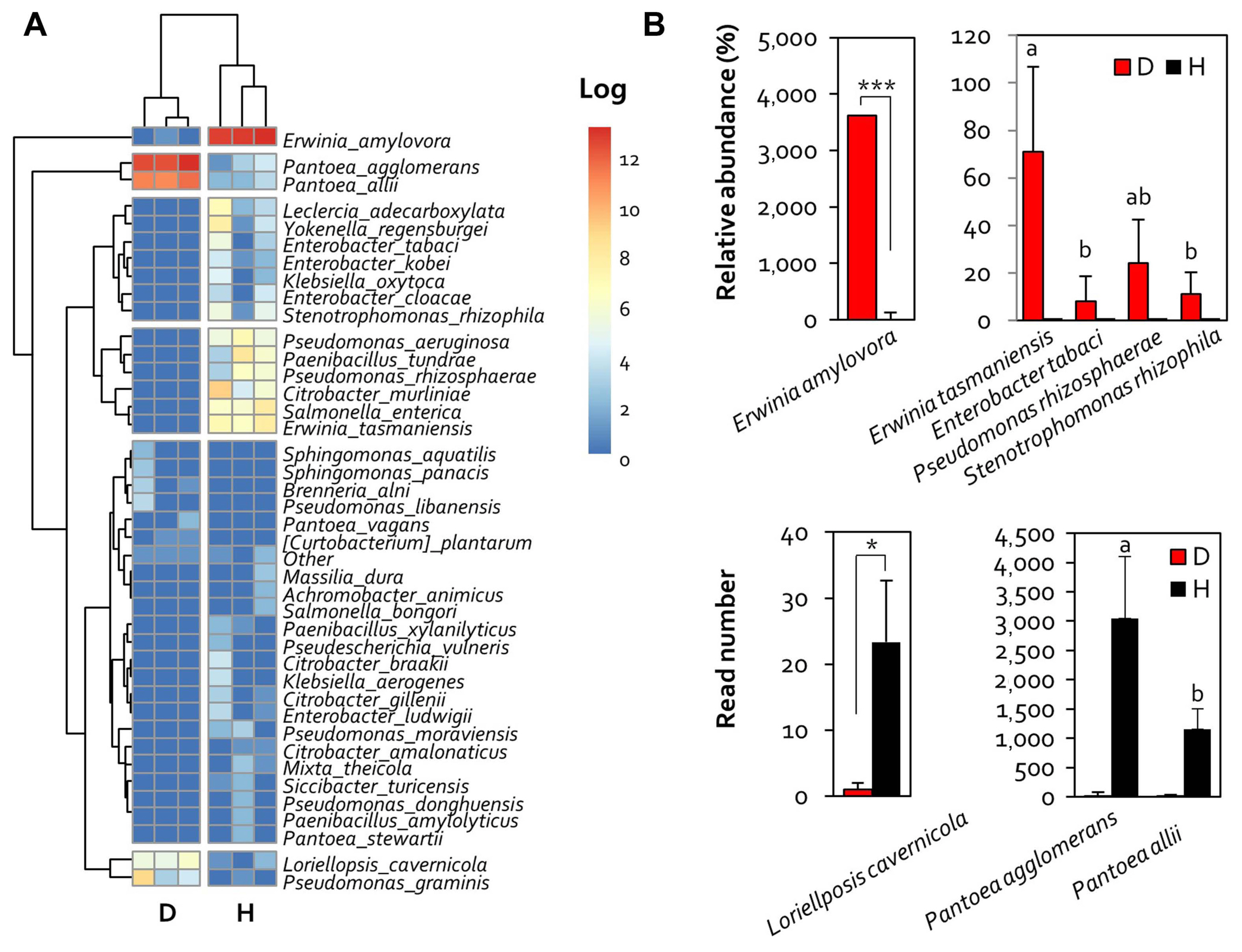
Differentially abundant bacterial communities were identified in diseased apple flowers. Family-level bacterial community structure analysis revealed that the majority (Ōēź 99%) of bacteria belonged to the Erwiniaceae family in all samples (diseased and healthy) (Fig. 1B). This suggests that the Erwiniaceae are closely correlated to the host flower.
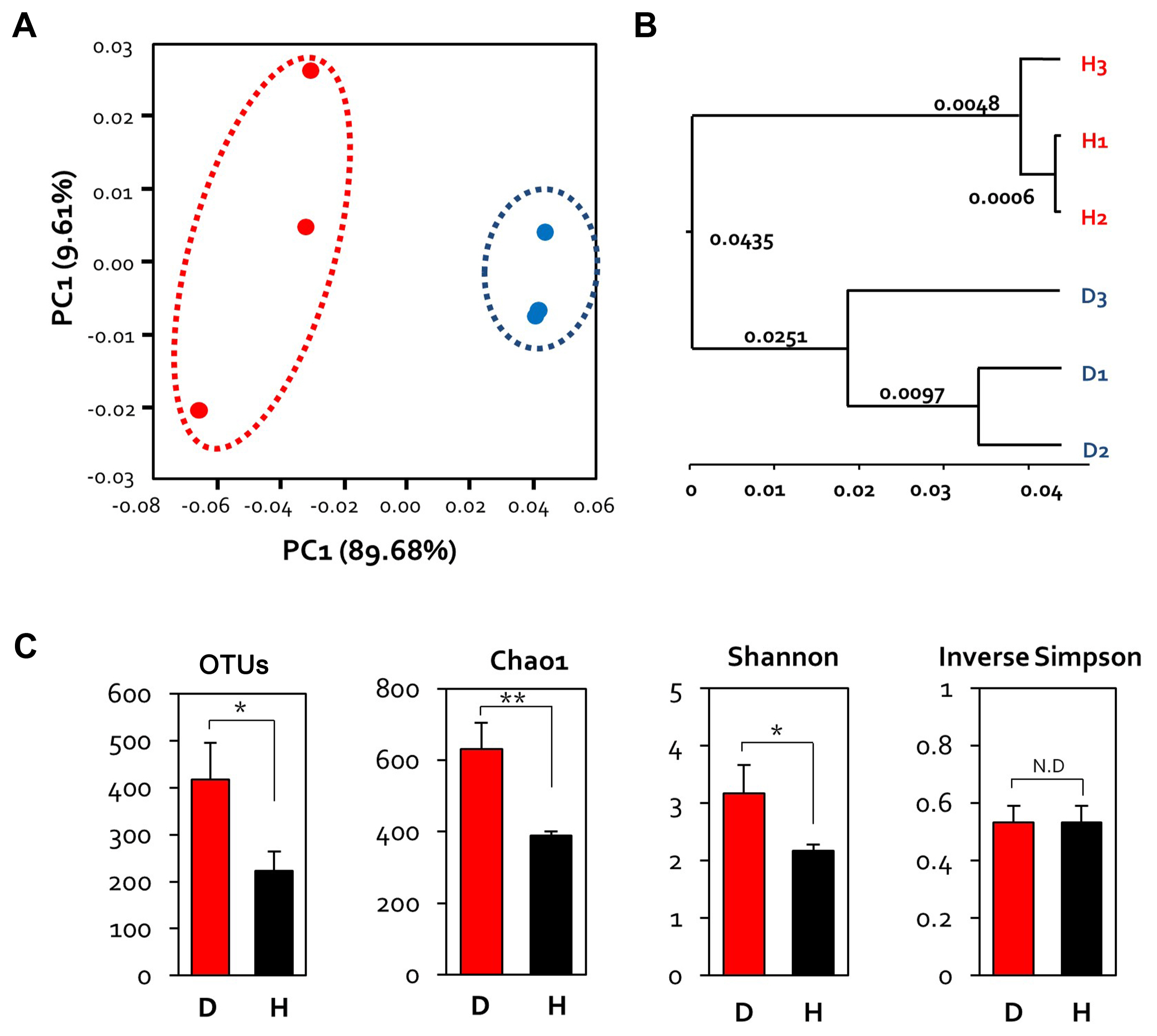
Principal coordinate analysis, based on the weighted UniFrac dissimilarity index, showed that the diseased and healthy samples formed separate clusters (Fig. 2A). The PCo1 and PCo2 axes accounted for 89.68% and 9.61% of the variance, respectively. The distance among samples within each group was variable; healthy flowers clustered closer together than the diseased flowers (Fig. 2A). This suggests that E. amylovora infection alters the microbial community of apple flowers, and these changes varied among individual flowers.
In order to confirm hierarchical clustering, it can be seen that grouping differently between diseased and healthy flowers is confirmed through unweighted pair group method with arithmetic mean tree (Fig. 2B). Alpha diversity analysis was performed based on richness and evenness indices. The bacterial evenness in the sample was estimated with the Shannon and Inverse Simpson indices. Bacterial richness showed no statistical significance, but diseased samples (OTU 400, Chao1 600) showed a higher than healthy samples (OTU 200, Cha1 400). On the other hand, values of the evenness index of diseased and healthy samples were similar (Fig. 2C).
Furthermore, microbial community analysis of each sample was performed at the species level. The results showed that E. amylovora was the most abundant bacterial species in the diseased sample (90% of all bacteria). On the other hand, Pantoea agglomerans and Pantoea allii were the most abundant bacteria (70% and 25%, respectively) in healthy samples (Fig. 3A). In addition, the abundance of Erwinia tasmaniensis, Pseudomonas, Salmonella, and Citrobacter was higher in diseased samples, while that of Lorielloppsis cavernicola was higher in healthy samples (Fig. 3B). These results indicated that E. amylovora infection diversified the microbial community of apple flowers, and the diversity of the microbial community was increased by the introduction of new bacteria rather than the existing inhabiting bacteria.
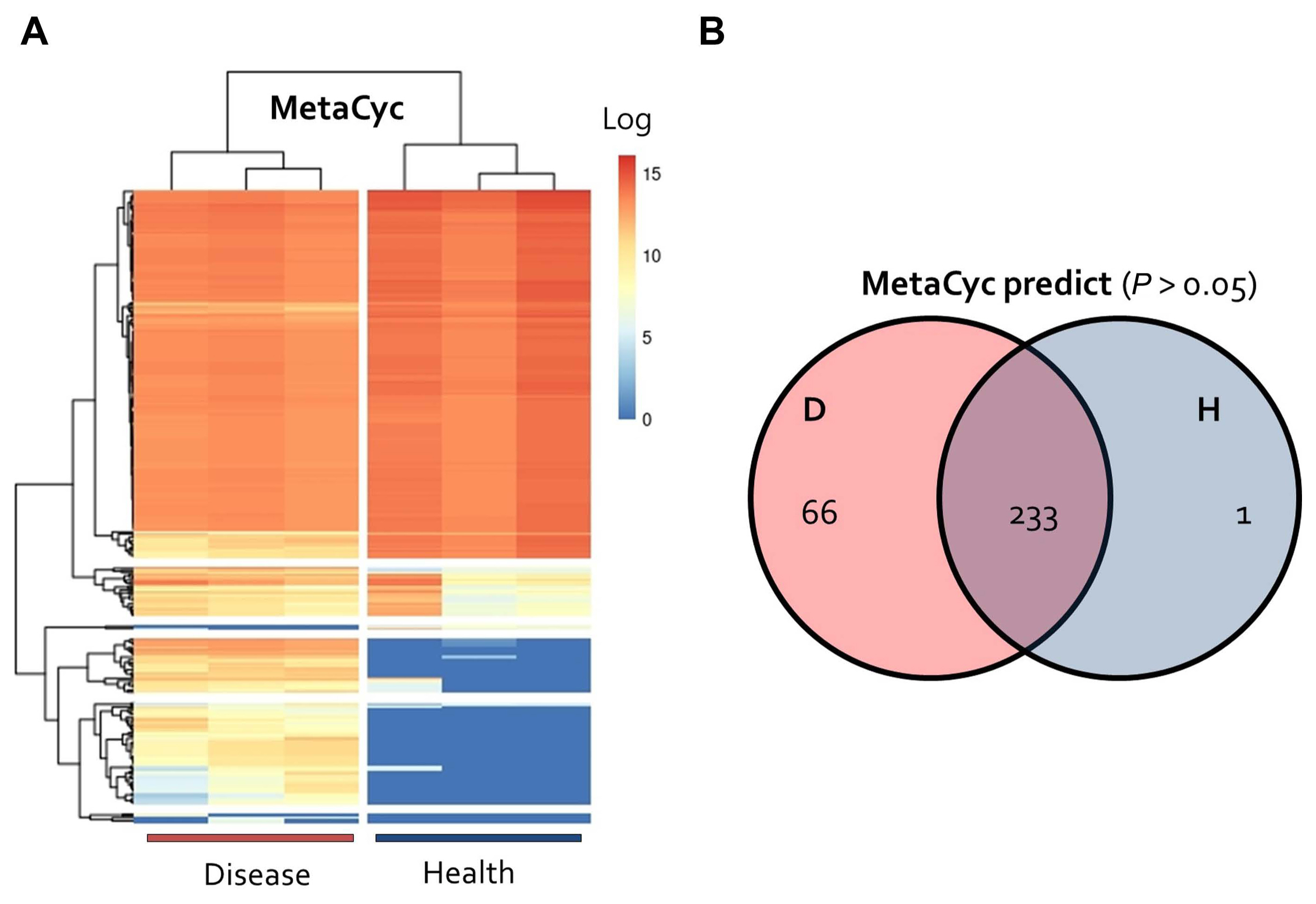
Our results showed that E. amylovora infection had a significant effect on the formation of microbial communities in apple flowers. The results of microbial community analysis were examined using the PICRUSt2 tool (Douglas et al., 2020) to determine functional genes (Fig. 4). Clustering analysis of 300 predicted metabolic MetaCyc pathways revealed the greatest variability between diseased and healthy samples, and the results showed that certain metabolic pathways were specific to the onset of the disease (Fig. 4A).
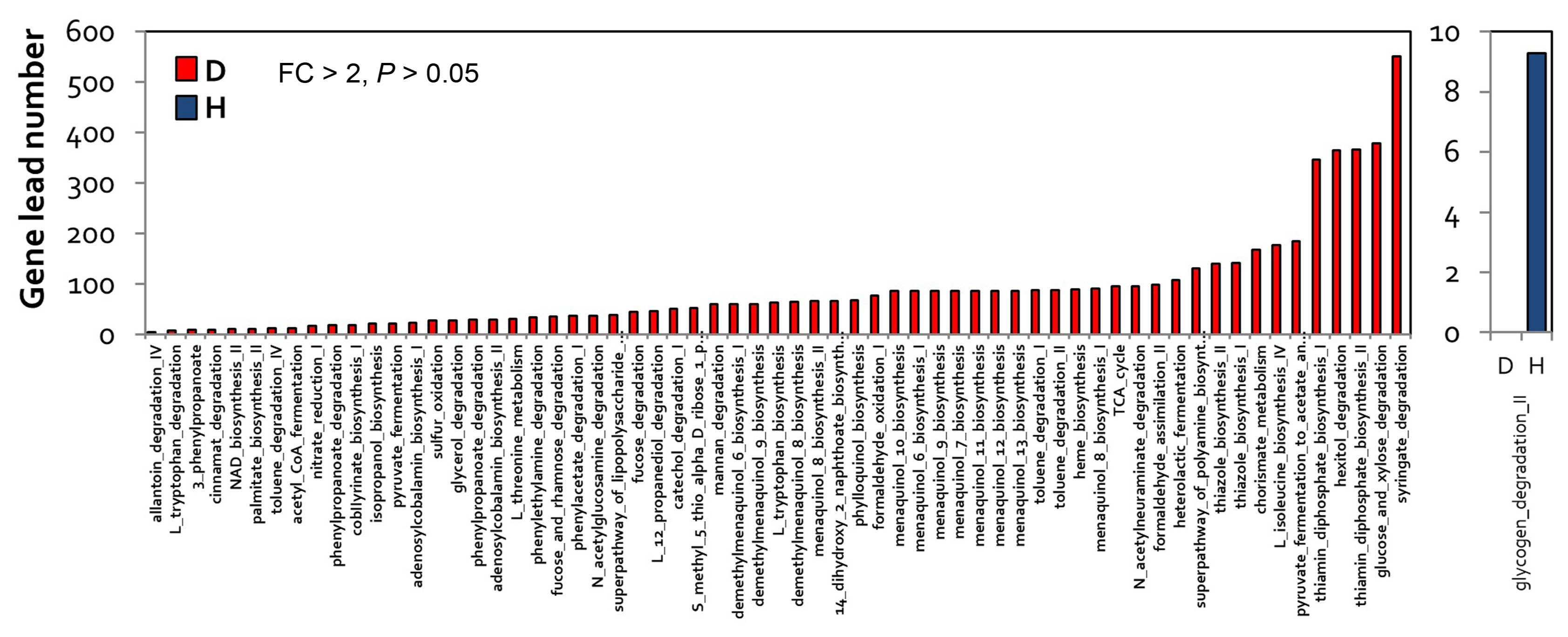
A total of 66 pathways (P < 0.05) specific to diseased samples were higher than one pathways (P < 0.05), which is a health-specific pathway (Fig. 4B). Analysis of the functions of these 66 pathways showed that pathways related to syringate, glucose, xylose degradation, and thiamin diphosphate biosynthesis were specifically enriched in diseased samples, whereas those related to glycogen degradation were specifically enriched in healthy samples (Fig. 5). In addition, pathways related to RR-butanediol, 2,3-butanediol, gallate, and catechol degradation were more highly enriched in diseased samples than in healthy samples (FC > 2, P < 0.05).
In bacteria, 2,3-(RR)-butanediol, a secondary metabolite, is produced during the process of pyruvate metabolism (Chen et al., 2020; Effantin et al., 2011; Venkataraman et al., 2014). In healthy samples, pathways related to glycogen, glycol, D-galactarate, and D-glucarate degradation were highly enriched (Supplementary Table 1). Overall, the analysis of microbiome and functional genes characterizes changes in microbial communities and functional genes caused by E. amylovora infection in apple flowers.
In this study, we compared the microbial communities of healthy and fire blight-infected apple flowers at the species level using the PacBio sequencing technology. The results showed that the microbial community of healthy flowers was predominantly composed of Erwiniaceae species. This result is consistent with the recently reported 16S rRNA (V3-V4) MiSeq data of apple flowers treated with E. amylovora (Cui et al., 2021).
This simplified microbiome in healthy flower is susceptible to multiple environmental changes and microbial infections, and suggests the possibility of dysbiosis in microbial communities (Chen et al., 2020). Thus, simplification of the microbiome implies low resistance to infection by bacteria, including E. amylovora. The results of this study showed that infection by E. amylovora increased the microbiome species diversity of apple flowers. The increase in the abundance of Erwinia tasmaniensis, Enterobacter tabaci, and Pseudomonas rhizosphaerae in diseased samples suggests the possibility of colonization after disease outbreak.
In addition, analysis of the microbial community at the species level confirmed that Pantoea species were the most predominant in healthy flowers. It has been shown that Pantoea species can grow at high sucrose concentrations (Pusey, 1999), suggesting that the sugar content of flowers is a major factor affecting the formation of microbial communities. In addition, the genus Pantoea belongs to the Erwinaceae family, similar to E. amylovora, and the pathogen also exhibits active sugar metabolism. Thus, we speculate that apple flowers provide the optimal environmental conditions for pathogen survival and infection.
Furthermore, functional gene analysis showed that the genes related to sugar metabolism were significantly enriched in the diseased flower samples. These results confirm that the presence of sugar molecules in flowers is closely related to the occurrence and proliferation of E. amylovora. In addition, we showed that the 2,3-butanediol related gene was significantly increased in diseased flowers. This result is consistent with the studies on volatile compounds, which are altered by fire blight. A previous study showed that 2,3-butanediol was detected only in plants inoculated with E. amylovora, and not in plants treated with Pseudomonas species (Cellini et al., 2018).
However, we could not determine whether the apple flower microbiome plays a causal role in the development of fire blight, or whether the presence of fire blight simply reflects an established infection with E. amylovora. Therefore, it is important to further explore the possible effects of the apple flower microbiome on fire blight-infected plant phenotypes, host immunity, and host-pathogen interactions. Furthermore, studies on the plant microbiome showed that the manipulation of the entire rhizome and lateral microbial community affects plant resistance and immunity as well as disease and infection (Hacquard et al., 2017). These findings suggest that manipulation of the apple flower microbiota could be used to inhibit plant disease. Thus, a species-level understanding of the apple flower microbiome may offer new opportunities for controlling fire blight.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print