Bacterial Community and Diversity from the Watermelon Cultivated Soils through Next Generation Sequencing Approach
Article information

Abstract
Knowledge and better understanding of functions of the microbial community are pivotal for crop management. This study was conducted to study bacterial structures including Acidovorax species community structures and diversity from the watermelon cultivated soils in different regions of South Korea. In this study, soil samples were collected from watermelon cultivation areas from various places of South Korea and microbiome analysis was performed to analyze bacterial communities including Acidovorax species community. Next generation sequencing (NGS) was performed by extracting genomic DNA from 92 soil samples from 8 different provinces using a fast genomic DNA extraction kit. NGS data analysis results revealed that, total, 39,367 operational taxonomic unit (OTU), were obtained. NGS data results revealed that, most dominant phylum in all the soil samples was Proteobacteria (37.3%). In addition, most abundant genus was Acidobacterium (1.8%) in all the samples. In order to analyze species diversity among the collected soil samples, OTUs, community diversity, and Shannon index were measured. Shannon (9.297) and inverse Simpson (0.996) were found to have the highest diversity scores in the greenhouse soil sample of Gyeonggi-do province (GG4). Results from NGS sequencing suggest that, most of the soil samples consists of similar trend of bacterial community and diversity. Environmental factors play a key role in shaping the bacterial community and diversity. In order to address this statement, further correlation analysis between soil physical and chemical parameters with dominant bacterial community will be carried out to observe their interactions.
Microbial communities are group of microorganisms that are inhabited in soil sharing a common space. Soil microbes may directly or indirectly affect the soil properties. Bacteria, actinomycetes, fungi, algae, and protozoa are the microbes found diverse in soil (Subba Rao, 1999). Soil bacteria could fix nitrogen as a result released oxygen into the atmosphere (Canfield, 2014; Subba Rao, 1999) which affects soil structure and fertility. Soil microbes has vital role in the soil environment. Change of soil environment is often linked with the variation of microbial populations (Kuang et al., 2018). Microbe’s diversity in soil plays a major role in maintaining healthy soil because these microbes are engaged in many vital functions like crucial cycles of C, N, P, formation of soil, removal of toxin, and so on (Fatima et al., 2014). Microbial composition and diversity are hugely known as prime factors in processing ecological activities (Bauman et al., 2013; Hernandez-Raquet et al., 2013). Numerous studies have found significant effects of biotic and abiotic factors on underground microbial composition and land management (Jangid et al., 2008).
Recently several studies have been focused in soil microbial community to modify and better use to promote plant health (Li et al., 2017). Soil microbiome analysis can predetermine whether plants are resistant or susceptible to diseases. Variation in soil microbiome composition and functioning can determine the reaction of plant-pathogen interaction under natural field conditions (Wei et al., 2019). Watermelon (Citrullus lanatus) is a major cucurbitaceous crop widely grown in tropical and subtropical regions across the world (Chomicki and Renner, 2015). Recently, plants have given account a holobiont, a unit representing both the host and its associated microbiome (Vandenkoornhuyse et al., 2015). Particularly, most roots and stem colonizing microorganisms seem to have originated from the rhizosphere while some are able to move systematically within the plant (Rosenblueth and Martinez-Romero, 2006). However, some microorganisms are restricted to belowground parts of plants (Compant et al., 2011). Such, plant-associated microbiome is highly diverse and holds a wide range of taxa. In order to study those microorganisms associated with plant parts and soil, composition and diversity analysis of those microorganisms is utmost.
Previous studies on the diversity of microbial communities rely on the isolation from soil sample using media selective technique for their identification morphologically. These media-based techniques for microbial community’s diversity study have been lead to biased method due to the unculturability of many microbes. Some soil microbes need specific media to grow in peculiar conditions. It has been reported that only 1% of soil microbes can be cultured in the laboratory using traditional culture techniques (Marchesi et al., 1998). In order to address these issues, microbiologists have adopted culture-independent techniques which include molecular biology based methods which consists of extracted DNA from soil samples is subjected to PCR amplification (Roh et al., 2006). These methods provide the detailed insights of species richness, evenness, and composition of bacterial community.
In order to provide insights in soil microbial activity and diversity, soil microbiome is the recently used technique which provides soil microbial community structure by using next generation sequencing (NGS) technology. This sequencing technology has shown a step way change in facilitating the characterization of soil prokaryotic and eukaryotic microbes (Bates et al., 2013; Fierer and Jackson, 2006; Ramirez et al., 2018; Tedersoo et al., 2014; Wu et al., 2011). Such sequencing technology for microbial community analysis uses ribosomal gene fragments from shotgun metagenomics (Guo, 2016). Microbiome research has crucial applications for understanding and manipulating ecosystem processes such as organic matter turnover, nutrient cycling, and the development or inhibition of soil pathogens (Philippot et al., 2013). Research in microbiome area has great ability to know the impacts of soil microbes on the productivity of natural plant communities and agroecosystems (McPherson et al., 2018).
The information regarding the bacterial community composition and diversity of watermelon cultivated soils in different geographical locations in Korea is limited and scarce. In order to address this, we have conducted this study with the following objectives: (1) to provide the insight information to researchers to describe the bacterial diversity present in soils in different geographical locations where the watermelon is cultivated, (2) to understand the soil surface ecosystem response to bacterial communities in varying environmental conditions.
Materials and Methods
Field site description and soil sampling
The study was conducted on August 2020 in South Korea. Detailed geographical coordinates and sampling locations have been presented in Table 1 and Supplementary Fig. 1. A total of 92 soil samples were taken from the use of soil auger (0–15 cm depth) from watermelon cultivated soils. Soil samples were collected in zipper bag with proper labeling. Each sample was placed in a plastic zipper bag and thoroughly homogenized before analysis. In the laboratory, the samples were sieved through a 2-mm mesh to remove roots and other debris and subsequently divided into two parts. Soil samples were kept at −20C for the subsequent DNA extraction and molecular analysis of community composition.
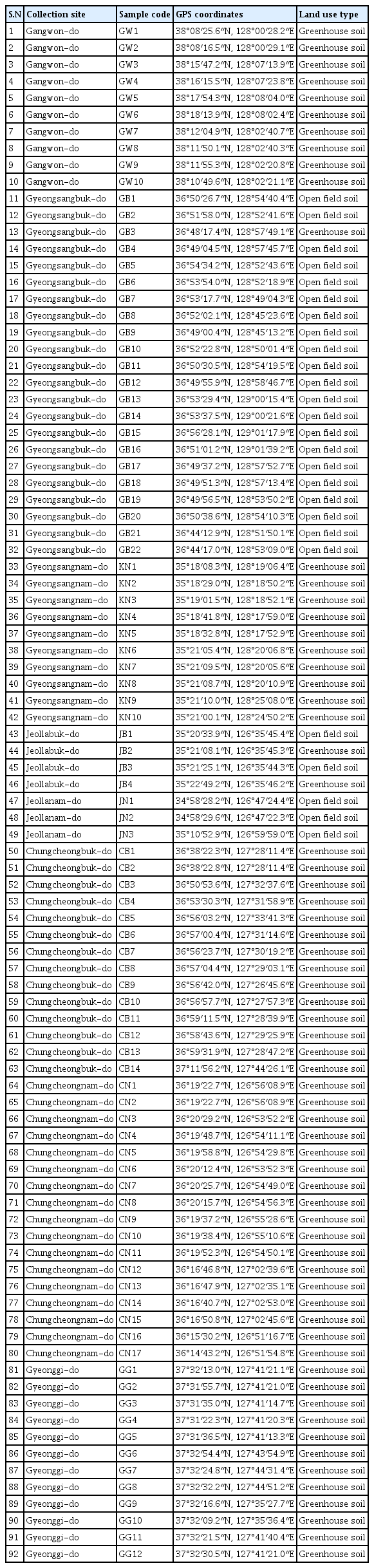
Soil samples collection sites with their GPS coordinates
Soil DNA extraction
Soil DNA extraction was carried out from the watermelon cultivated soils to sterilized buffer (NaCl, 4.25 g; KH2PO4, 0.15 g; Na2HPO4, 0.3 g; MgSO4 0.1 g; gelatin, 0.05 g), mixed, and allowed to stand for 10 minutes. Soil solution was extracted using Fast DNA Spin Kit for soil (MP, Biomedicals, Seoul, Korea): Fast DNA SPIN Kit for soil according to the extraction method. A quantitative polymerase chain reaction (qPCR) amplification was conducted after the total DNA extraction from the soil samples. A qPCR amplification of the prokaryotic microbes (16S rRNA genes V4 region) was performed using the forward primer 515F (5′-GTGYCAGCMGCCGCGGTAA-3′) and the reverse primer 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Bates et al., 2011). Amplicons were sequenced on Illumina-MiSeq platform (Illumina Inc., San Diego, CA, USA) at SHBIO Technology (Shanghai, China).
Analysis of sequencing data for bacteria community composition
Raw sequences received after PCR amplicon sequencing were analyzed and processed mainly in three steps: data trimming, taxonomy assignment and normalization, and diversity analysis. Briefly, raw sequences were filtered using FastQC run using the software fastqc v0.11.2 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). Forward and reverse direction of fastQC files were assimilated from software vsearch v2.10.3 (https://www.github.com/torognes/vsearch), followed by filtering of the chimeric sequences. This process leads to the removal of unassembled and low-quality sequences with >97% similarity could be clustered into operational taxonomic units (OTU). OTU and the representative sequences for each OTU were constructed using QIIME (Quantitative Insights into Molecular Ecology) software (version 1.17) package (Jiang et al., 2017) to annotate the taxonomic information for each representative sequence.
Analysis of soil bacterial diversity
Alpha and beta diversity analyses were carried out based on the normalized OTU data. Alpha diversity indices, including Chao1, Shannon, and Simpson indices, were calculated with QIIME (version 1.7.0) (Caporaso et al., 2010) to assess the richness and species diversity for each soil sample of different geographical locations. The beta-diversity patterns of the bacterial communities (Bray-Curtis dissimilarities) (Sokal and Michener, 1958) were characterized using principal coordinate analysis (PCoA) and the unweighted pair-group method with arithmetic mean (UPGMA) clustering based on unweighted UniFrac distances. The unweighted Uni-Frac (UUF, qualitative measure) assesses the presence or absence of taxa and is, thus, more sensitive to rare taxa.
Results and Discussion
Bacterial community composition across the different geographical locations
In order to find the bacterial fruit blotch causing pathogen, Acidovorax citrulli from the soil samples collected from different locations cultivated with watermelon plants NGS of the hypervariable V1 and V4 regions of bacterial 16S ribosomal RNA was performed. Results showed diverse bacterial community composition in all the locations. Abundancy analysis results showed that, 6 (Proteobacteria, Acidobacteria, Actinobacteria, Bacteroidetes, Gemmatimonadetes, and Verrucomicrobia) bacterial phylum dominantly presents in all of the 92 soil samples (Fig. 1). However, Proteobacteria, Bacteroidetes, Acidobacteria, Gemmatimonadetes, Verrucomicrobia, Actinobacteria, Chlorofexi, Firmicutes, Ignavibacteriae, Planctomycetes, and Nitrospirae were the dominant phylum detected in two different land use type soils (open field and greenhouses) from all the provinces. Among these, Proteobacteria was the most abundantly dominant bacterial phylum in all the soil samples. Soil samples from Gangwon-do, comprise about 50%, 17%, and 10% of Proteobacteria, Bacteroidetes, and Acidobacteria, respectively (Supplementary Fig. 2). Soil samples from Gyeongsangbuk-do also mostly dominated by Proteobacteria, Acidobacteria, and Bacteroidetes with around 44%, 27%, and 22%, respectively (Supplementary Fig. 3). In case of soil samples from Gyeongsangnam-do, Proteobacteria, Actinobacteria and Firmicutes were the most dominant in the ratio about 48%, 35%, and 14%, respectively (Supplementary Fig. 4). Dominant phylum coverage of the bacteria from the soil of Jeollabuk-do were Proteobacteria (32–51%), Bacteroidetes (11–22%), and Acidobacteria (8–10%) (Supplementary Fig. 5). Soil samples from Jeollanam-do hold Proteobacteria, Acidobacteria, and Bacteroidetes as the most dominant bacteria phylum with the abundancy ratio of about 37%, 22%, and 14%, respectively (Supplementary Fig. 6). Abundance ratios of around 63%, 54%, and 10% of Bacteroidetes, Proteobacteria, and Acidobacteria was obtained from soil samples of Chungcheongbuk-do (Supplementary Fig. 7). Soil samples from Chungcheongnam-do were highly accounted for Proteobacteria, Bacteroidetes, and Acidobacteria with the dominant ratios of around 43%, 20%, and 18%, respectively (Supplementary Fig. 8). Moreover, soil samples from Gyeonggi-do consisted of about 46%, 20%, and 10% of Proteobacteria, Bacteroidetes, and Actinobacteria, respectively (Supplementary Fig. 9). Monitoring soil bacterial communities in soils from different geographical conditions is a major challenge. It helps implement a reliable sustainable management of soils in agriculture (Bender et al., 2016). Effective knowledge regarding the geographical distribution of microbial communities is critical to better understand the ways soils under different climate conditions (Plassart et al., 2019). In addition, pattern of bacterial diversity is markedly different from that of plant species and most soil bacteria and may alter with environmental and geographical distance (Wang et al., 2015). However, primary factors affecting microbial spatial distribution are still under debate (Lauber et al., 2009). The distribution of soil bacterial community can be altered by various factors like; soil properties, litter quality, root exudates, and also by temperature and precipitation in different geographical conditions (Nielsen et al., 2010). In this study, we observed slight variation in dominant bacterial composition across the different geographical locations (Fig. 1). However, factors affecting such variations in bacterial composition in different geographical locations need to be studied further.
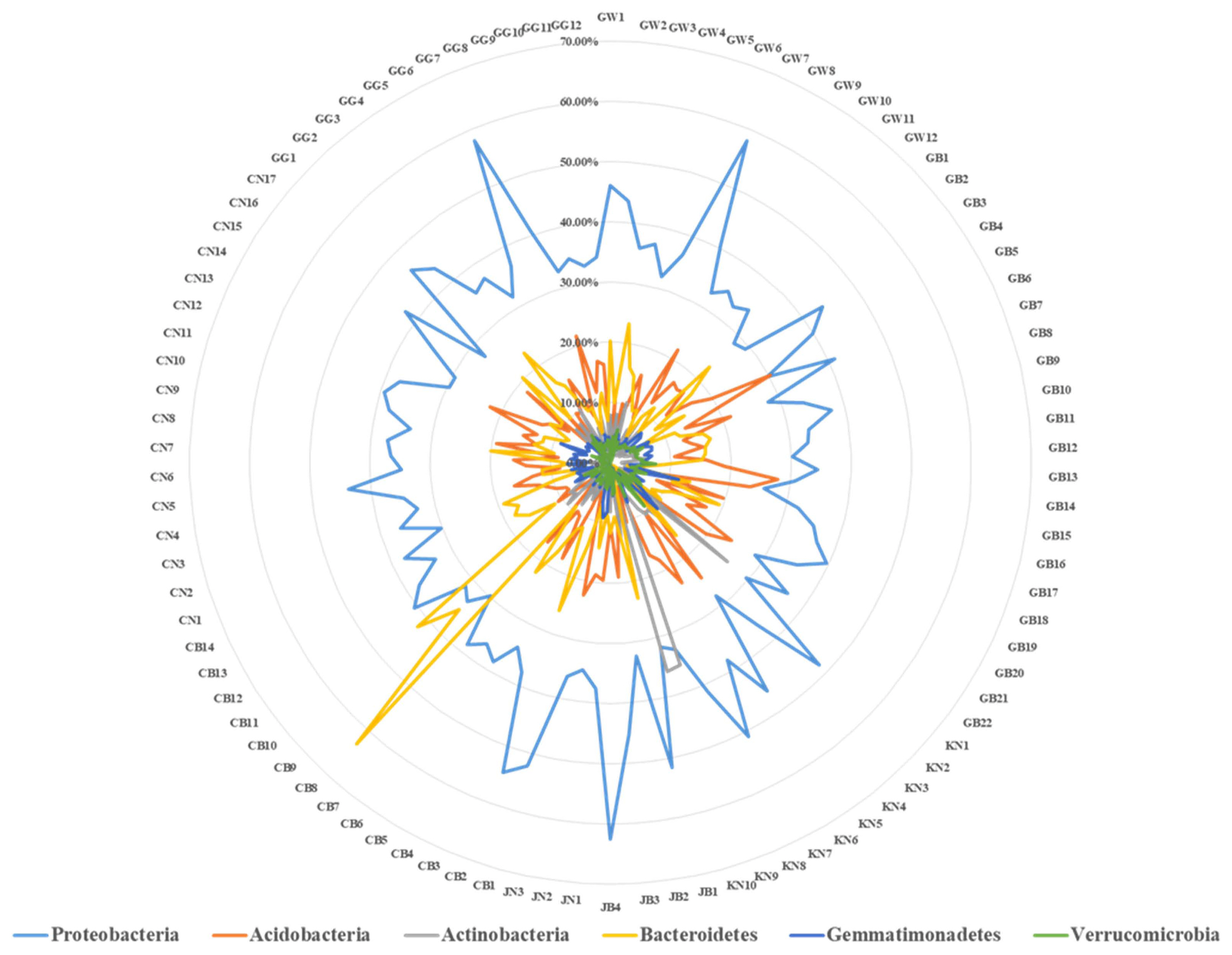
Top most dominant phylum along with the abundance ratio detected across all of the 92 soil samples. GW, Gangwon-do; GB, Gyeongsangbuk-do; KN, Gyeongsangnam-do; JB, Jeollabuk-do; JN, Jeollanam-do; CB, Chungcheongbuk-do; CN, Chungcheongnamdo; GG, Gyeonggi-do.
Moreover, in this study, we are also trying to assess whether our collected watermelon cultivated soil samples consist A. citrulli or not from various geographical locations. In this regard, we could not observe any A. citrulli isolate in our studied soil samples as this bacterial pathogen is a seed-borne disease affecting the embryo and endosperm of seeds (Rahimi-Midani et al., 2018). Furthermore, previous reports suggest that, A. citrulli actively colonize and move through xylem vessels then reach fruits and seeds (Bahar et al., 2009) and the chance of moving this pathogen into soil is very rare except by the other external influencing factor. Not detection of A. citrulli in our studied soil samples might be due to the above mentioned reasons.
Bacterial diversity across the different geographical locations
Bacterial diversity was analyzed in soils of different provinces. Alpha diversity was assessed using Chao1, Shannon indices, and beta diversity was analyzed using PCoA. The OTU in all the analyzed soil samples was in range of 276 to 2216. OTU is an operational definition used to classify groups of closely related individuals and ‘OTU’ is simply the group of organisms currently being studied. The number of OTUs in this study was found lowest from the greenhouse soil of Jeollabuk-do (JB4) and highest from greenhouse soil of Gyeonggi-do (GG4) (Fig. 2). Bacterial diversity according in land use types also was investigated in this study. Twenty-six soil samples were from open-field (GB1-GB22, JB1 and JB3, JN1-JN3) and 66 were from greenhouse (GW1-GW10, KN1-KN10, CB1–14, CN1-CN17, and GG1-GG12). OTUs from open-field soil was recorded highest in GB12 and lowest in JB1 (Supplementary Fig. 10). Soils from greenhouse of GG4 was the highest OTUs as compared with other greenhouse soils (Supplementary Fig. 11). For several years, OTUs have been the most commonly used units of diversity, especially when analyzing small subunit 16S for prokaryotes marker gene sequence datasets (Sommer et al., 2017). In order to relate this with our study, 16S rRNA metagenomics sequencing revealed the JB4 with the lowest OTUs and GG4 with the highest OTUs. In total soil samples, Shannon and inverse Simpson diversity indices were found lower in JB4 (Shannon, 3.53; Inverse Simpson, 0.73) and highest in GG4 (Shannon, 9.29; Inverse Simpson, 0.995) (Fig. 3). Moreover, diversity indices (Shannon and inverse Simpson) according to land use types were also observed. Results suggest that, among the open field soils, diversity was higher in GB12 and lower in JB1 (Supplementary Fig. 12). Whereas, among the greenhouse soils, diversity was higher in GG4 and lower in JB1 (Supplementary Fig. 13). The Shannon diversity index represents the microbial community richness, indicating the more abundant species in the community (Hill et al., 2003). Microbial diversity has been closely associated to soil health. Microbial community diversity enhances the well-functioning of the soil ecosystem. Rao 1999 reported that high microbial diversity is an indicator of a healthy soil, while low microbial diversity indicates a stressed condition of soil. Moreover, the microbial diversity of soil is the most crucial component that reflects the quality and stability of soil (Cassán and Diaz-Zorita, 2016) and has the ability to set various non-biological indicators. Hence, the exploration of soil microbial diversity is critical factor for the estimation of health and fertility of soil using high-throughput DNA sequencing technology (Roesch et al., 2007). In this study, however, all the soils samples were from watermelon cultivated soils, bacterial community diversity were found more diversified. Bacterial species richness and diversity (OTUs, Chao1, Shannon, Inverse Simpson) was assessed lowest in soil of Jeollabukdo (JB4) and highest in soil of Gyeonggi-do (GG4).
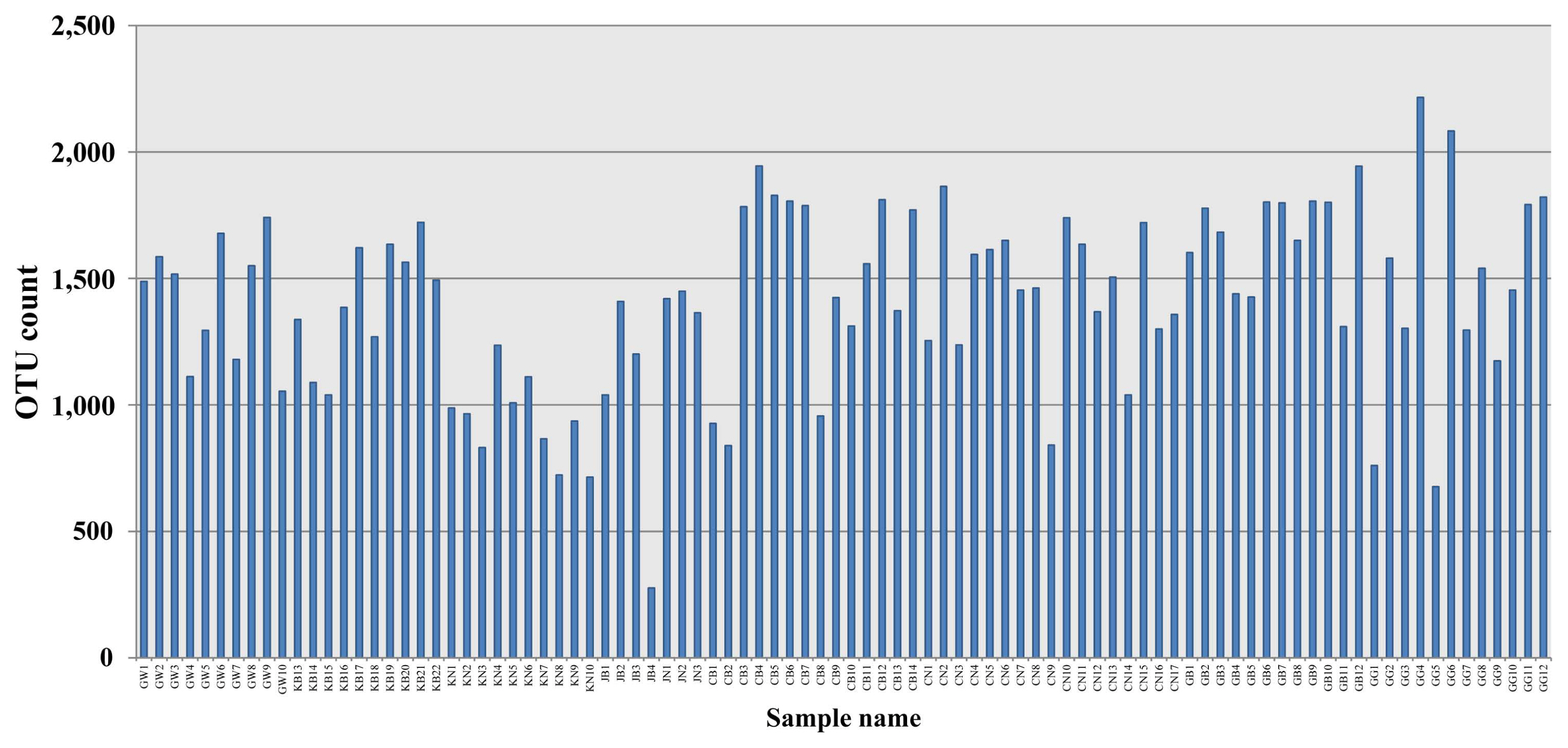
Operational taxonomic units (OTUs) of bacterial community obtained from total 92 soil samples from watermelon cultivated soil across the different provinces. X-axis indicates soil samples from various geographical locations: GW, Gangwon-do; KB and GB, Gyeongsangbuk-do; KN, Gyeongsangnam-do; JB, Jeollabuk-do; JN, Jeollanam-do; CB, Chungcheongbuk-do; CN, Chungcheongnamdo; GG, Gyeonggi-do.
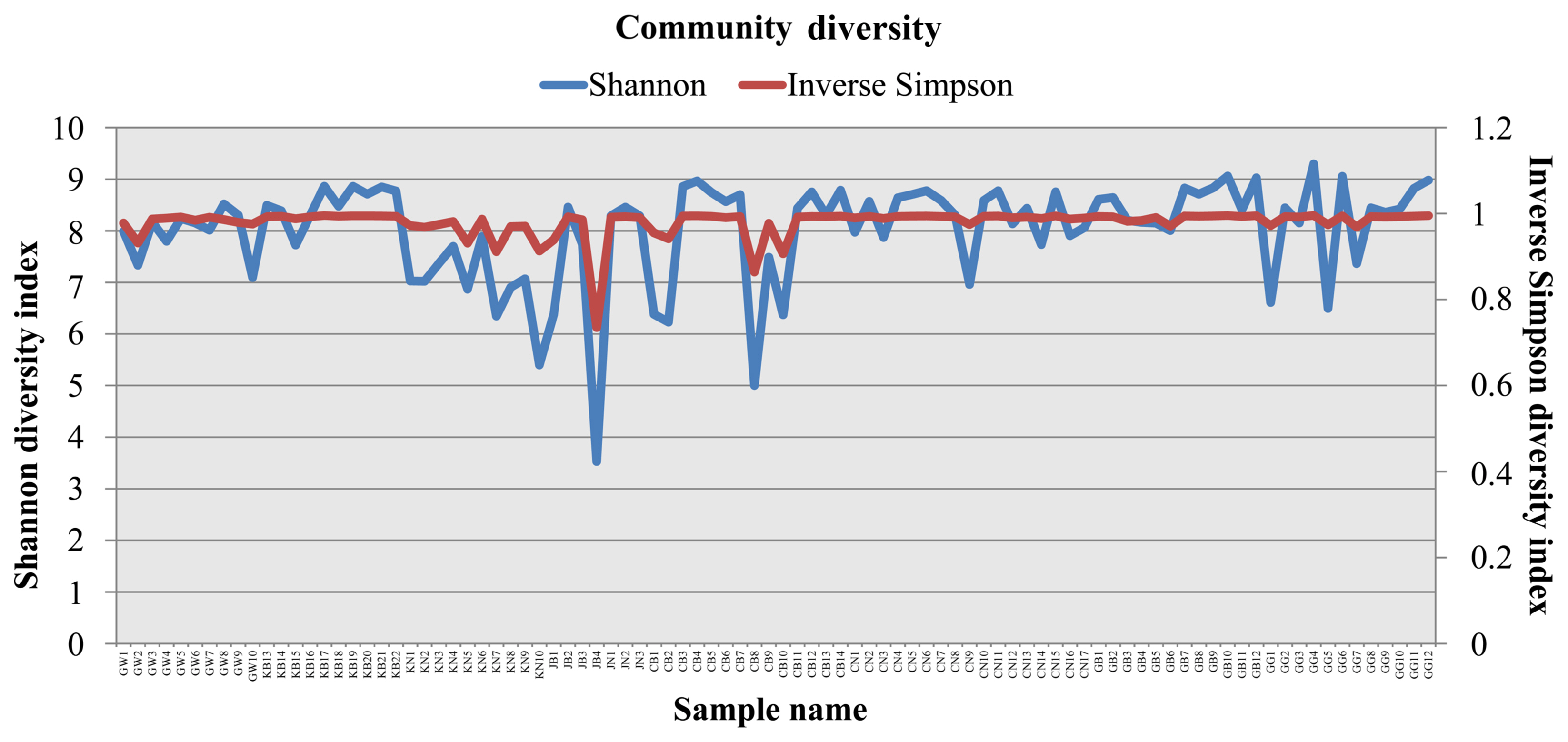
Bacterial community richness and diversity obtained from total 92 soil samples from watermelon cultivated soils across the different provinces. X-axis indicates soil samples from various geographical locations: GW, Gangwon-do; KB and GB, Gyeongsangbuk-do; KN, Gyeongsangnam-do; JB, Jeollabuk-do; JN, Jeollanam-do; CB, Chungcheongbuk-do; CN, Chungcheongnam-do; GG, Gyeonggi-do.
Variation in bacterial community structure in different geographical locations
UPGMA and PCoA plot was used to visualize the variation in bacterial community structure in watermelon cultivated soils across the different geographical locations. The unweighted UniFrac (UUF, qualitative measure) provides the status of taxa which is more sensitive to rare taxa. The UPGMA tree in our study revealed that, bacterial communities from soil samples of same province lie in nearby clusters (Fig. 4). Qualitative measurement of bacterial community among different provinces mostly exhibited separate clade in UPGMA tree (Fig. 5). However, some bacterial community taxa of soil samples of same province showed slight variation as seen in the UPGMA tree (Fig. 5). Interestingly, unweighted and weighted unifrac tree showed that, only bacterial community from the soils of Gyeongsangnam-do (KN1-KN10) lies in same clade (Figs. 4 and 5). In contrast, bacterial community from the soils of Chungcheongbuk-do (CB1-CB14) were lying in different clade in both unweighted and weighted unifrac tree (Figs. 4 and 5). Measures of variation in microbial community composition in different soils were broadly performed by β-diversity. Examining the patterns of β-diversity (similarities/dissimilarities) across different land uses soils provides insights into the dynamics of microbial communities (Anderson et al., 2011). In our study, we have tried to examine the patterns of bacterial community composition in watermelon cultivated soils in different locations in South Korea. PCoA plot of weighted and unweighted unifrac tree exhibited significant variation in bacterial community structure among the land use types across different geographical locations (Figs. 6 and 7). Our study distinctly showed that the dominant bacterial community composition was slightly varied among the locations. Index for each bacterial community composition in respective geographical locations has been indicated in the PCoA graph of Figs. 6 and 7.
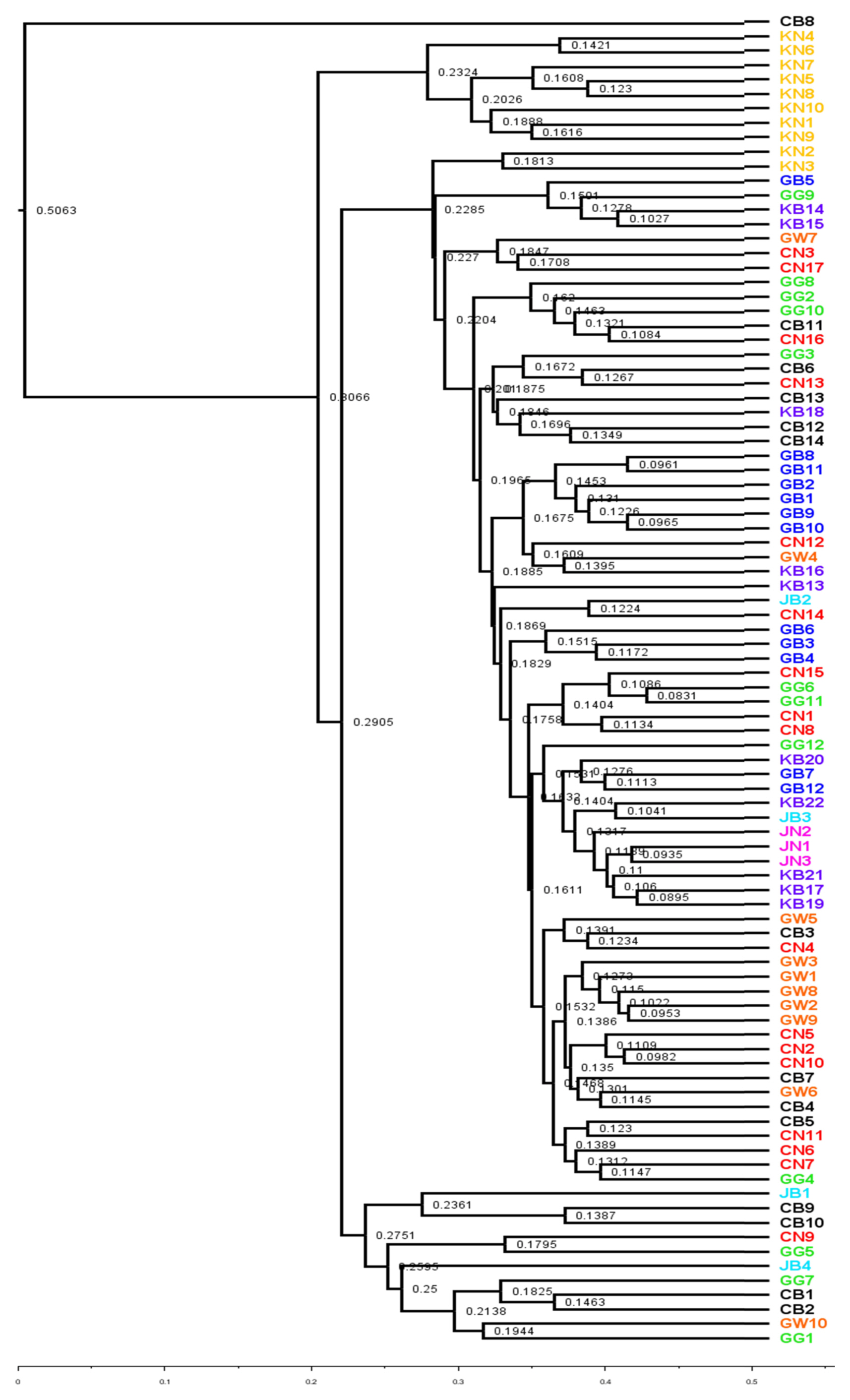
Weighted unifrac tree showing hierarchical clustering of average linkage of bacterial community from 92 soil samples.
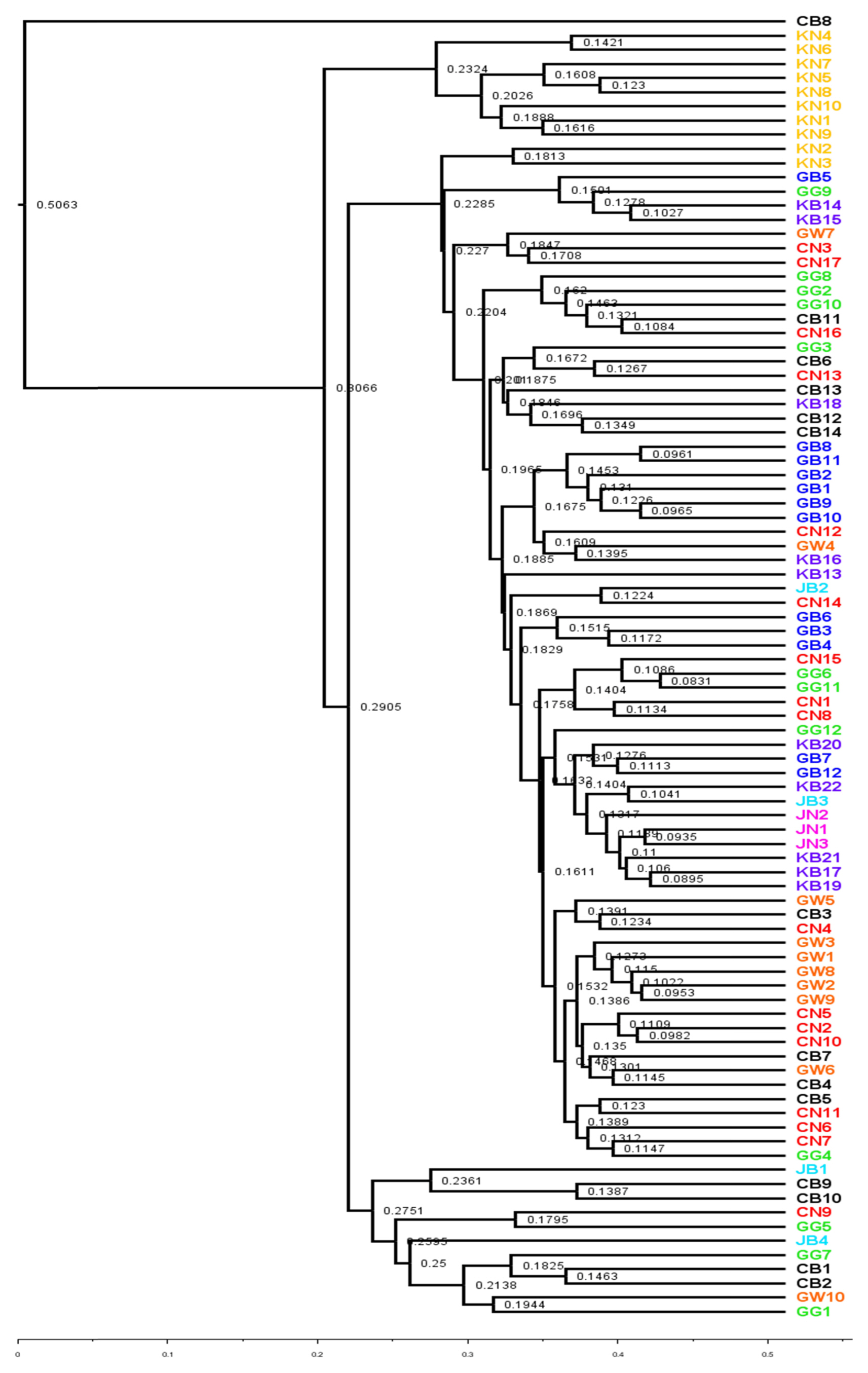
Unweighted unifrac tree showing hierarchical clustering of average linkage of bacterial community from 92 soil samples.

Principal coordinate analysis (PCoA) plot of weighted unifrac showing variation based on the group of the bacterial communities encountered across the soil samples. Different colored plots indicate the variation in bacterial community composition according to land use types and geographical locations.

Principal coordinate analysis (PCoA) plot of unweighted unifrac showing variation of the bacterial communities encountered based on the group across the soil samples. Different colored plots indicate the variation in bacterial community composition according to land use types and geographical locations.
Conclusion
The highest bacterial diversity was observed in the greenhouse soil from Gyeonggi-do province (GG4) and the lowest bacterial diversity was detected in the greenhouse soil from Jeollabuk-do province (JB1). We have found slight variation in bacterial diversity in open field soils and greenhouse soils of different provinces. However, bacterial community composition and diversity was somewhat similar from the soil samples of same land use types. Significant reason behind the variation in bacterial diversity is might be due to the soils from different locations and land use types. Alpha diversity indices (OTUs, Shannon and Inverse Simpson) and beta diversity indices (UPGMA tree, dissimilarity) are the key factors to determine overall diversity of microbes present in the soil. In this study, we observed watermelon cultivated soil samples from nearby area holds similar bacterial community composition and diversity as compare to different areas through UPGMA tree using distance matrix values. In contrast, we could not find the A. citrulli from our soil samples and further soil samples and watermelon plant samples will be analyzed in order to find the A. citrulli using NGS approach. However, further studies regarding the correlation of chemical attributes shaping the bacterial community in different land use type are further needed to draw the general trend. Thus, to the best of our knowledge, this is the first report regarding bacterial community composition and diversity from the watermelon cultivated soil in various geographical locations in South Korea. Also, we expect that the soil microbiome analysis could provide some biological indicators for the assessment of soil health and ecosystem services, but for this purpose, further studies with a holistic approach would be worthwhile.
Acknowledgments
This work was supported by Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, and Forestry (IPET) through Project No. 120088-05-1-SB010 funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA). This research has been worked partially with the support of a research grant of Kangwon National University in 2019 and 2020.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Electronic Supplementary Material
Supplementary materials are available at The Plant Pathology Journal website (http://www.ppjonline.org/).