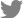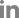The classification of the genus Pectobacterium, a causal agent of soft rot, has recently been revised due to its phenotypic and genotypic heterogeneity. This genus has been reclassified into 20 species: P. aroidearum (Par), P. aquaticum, P. atrosepticum (Pat), P. actinidiae (Pac), P. betavasculorum, P. brasiliense (Pbr), P. cacticida, P. carotovorum (Pca), P. fontis, P. odoriferum (Pod), P. parmentieri (Ppa), P. parvum (Ppv), P. peruviense, P. polaris (Ppo), P. polonicum, P. punjabense, P. quasiaquaticum, P. versatile (Pve), P. wasabiae, and P. zantedeschiae (Jee et al., 2020; Moussa et al., 2021; Pasanen et al., 2020; Portier et al., 2020). Pasanen et al. (2020) suggested a new species, Ppv, differentiated from Ppo with low average nucleotide identity (ANI) coverage and reported a type III secretion system (T3SS) of Salmonella pathogenicity island 1 (SPI-1). Among the 20 species, 8, (Pat, Pac, Pbr, Pca, Pod, Ppa, Ppo, and Pve) were isolated from various hosts in South Korea (Jee et al., 2020; Lee et al., 2014, 2017; Park et al., 1999). Due to the heterogeneous genome of Pectobacterium strains, new species may be observed upon further examination, as confirmed previously (Portier et al., 2020). We previously identified that Pca, Pod, Pbr, and Pve were the prevalent species isolated from potatoes in South Korea (Jee et al., 2020). This result was specific to South Korea because Pod is known as a soft rot pathogen that primarily causes the disease in vegetables.
Attempts have been made to understand pathogenic characteristics via whole-genome sequencing and comparative analysis of Pectobacterium (Arizala and Arif, 2019; Li et al., 2018, 2019). Although some species were sequenced incompletely, such as P. fontis and P. betavasculorum, at least one strain of the prevalent species has been completely sequenced at the whole-genome level. Pectobacterium demonstrates pathogenic factors, such as plant cell wall degrading enzymes (PCWDEs), secretion systems, and quorum sensing systems, which aid in the infection of host plants. PCWDEs include cellulases, proteases, and pectindegrading enzymes, including pectin lyases, pectin esterases, and polygalacturonases. Pectobacterium is known to effectively degrade plant cell walls using these enzymes. Type I secretion system (T1SS) and type II secretion system (T2SS) are used by pathogens for the extracellular secretion of enzymes. T1SS is known for one-step transport, while T2SS, located in the outer membrane, requires Sec or Tat secretion pathway to deliver the target protein to the periplasm (Green and Mecsas, 2016). First identified in 1994, T3SS could transfer toxic proteins acting as effectors directly into the host cell. T3SS is considered an important factor for pathogenicity as it aids gram-negative bacterial pathogens in infecting a variety of hosts, ranging from plants to humans. Type VI secretion system (T6SS) was first reported in 2006, and its apparatus resembles a phage structure (Bernal et al., 2018). T6SS targets eukaryotic and prokaryotic cells for host manipulation and interbacterial competition.
Jee et al. (2020) evaluated the phenotype of pathogenic factors according to temperature with Pectobacterium strains isolated in South Korea. To identify these pathogenic factors in the genome, seven strains from South Korean collection isolated by Jee et al. (2020) were selected as representatives for each species. We conducted complete whole-genome sequencing and comparative analysis with seven representatives of South Korean Pectobacterium. The strains isolated in South Korea for full genome sequencing are Pbr strain PCC21 and three Pac strains: KKH3, ICMP 19971, and ICMP 19972 (Lee et al., 2017; Park et al., 2012; Visnovsky et al., 2017). The objective of this study was to update the genome sequence information of Pectobacterium species that occur in South Korea and understand the pathogenicity factors associated with their genomes.
Materials and Methods
Bacterial strains and genomic DNA extraction
Seven bacterial strains were selected as the representative strains of the Pectobacterium species isolated in South Korea. The seven bacterial strains consisted of six species: Pca, Pod, Pbr, Pve, Ppv, and Ppa. Jee et al. (2020) isolated five strains from potato (Solanum tuberosum), P. carotovorum JR1.1 from radish (Raphanus sativus), and P. odoriferum JK2.1 from kimchi cabbage (Brassica rapa subsp. pekinensis). The seven strains were cultivated in Luria-Bertani medium (BD, Franklin Lakes, NJ, USA) at 28┬░C for 16 h. Genomic DNA of seven Pectobacterium strains was extracted using the G-spin Genomic DNA Extraction Kit (Intron Biotechnology, Seongnam, Korea) according to the manufacturerŌĆÖs instructions.
Whole-genome sequencing and annotation
The extracted DNA was examined via electrophoresis using 1.0% agarose to assess DNA quality. The complete genomes of seven Pectobacterium strains were sequenced at Phyzen Co. Ltd. (Seongnam, Korea) using the PacBio RS II platform with a Template Prep Kit from the SMART bell library. The constructed library was validated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). De novo assembly was performed using PBcR (ver. 8.3.rc2), SMARTdenovo (ver. 1.0.0), and Flye (ver. 2.7-b1587). The completeness of each genome was measured with Benchmarking Universal Single Copy Orthologs (BUSCO) score using bacteria_odb9 library. Gene prediction was performed using the NCBI Prokaryotic Genome Annotation Pipeline (Haft et al., 2018; Li et al., 2021; Tatusova et al., 2016). GenBank accession numbers were assigned to each strain in this study (Table 1).
Comparative genome analysis
All genome sequences of Pectobacterium were obtained from the NCBI database. To compare homology patterns among the seven Pectobacterium strains, genome sequences were aligned with the progressive Mauve algorithm using Mauve (ver. 2.4.0, http://darlinglab.org/mauve/) (Darling et al., 2004). ANIs were evaluated using the ANI/AAI-Matrix (http://enve-omics.ce.gatech.edu/g-matrix/), and in silico DNA-DNA hybridization (isDDH) values were calculated using the default settings of the Genome-to-Genome Distance Calculator (GGDC 3.0, http://ggdc.dsmz.de/ggdc.php) (Meier-Kolthoff et al., 2021). A pan-genome phylogenetic tree was constructed with the bacterial pan-genome analysis pipeline software (BPGA, https://iicb.res.in/bpga/) using recommended settings and MEGA7 program using neighbor-joining method with 1,000 bootstraps (Chaudhari et al., 2016). Thirteen reference species were retrieved from the GenBank database (Supplementary Table 1). For functional genomics analysis among the strains, the coding sequences (CDSs) were classified into 25 functional groups with reference to orthologous groups (EggNOG 4.5, http://eggnogdb.embl.de) (Powell et al., 2014).
CAZyme and pathogenesis-related gene analysis
Carbohydrate-active enzymes (CAZymes) of the seven Pectobacterium strains were analyzed using the dbCAN meta server (http://bcb.unl.edu/dbCAN2/) (Zhang et al., 2018). Pathogenesis-related genes, such as those encoding PCWDEs and type secretion systems, including T1SS, T2SS, T3SS, and T6SS from one to six in each genome, were identified and compared using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi), such as BLASTN, BLASTX, and BLASTP, and UniProt (https://www.uniprot.org/) with previously known genes of Pectobacterium (Li et al., 2018, 2019). Signal peptides and subcellular locations of proteins were identified using UniProt. For accuracy, each BLAST hit of an open reading frame was limited to over 40% query coverage, the lowest e-value, and the highest percent identity. This study identified three types of enzymes: cellulases, proteases including metalloproteases and serine proteases, and pectin-degrading enzymes including pectate lyases, pectin lyases, pectin esterases, rhamnogalacturonate lyase, and polygalacturonases. The motif of the putative effector of T6SS was identified using MEME (ver. 5.3.3, https://meme-suite.org) (Bailey et al., 2015).
Results and Discussion
Comparison of the whole-genome sequences of South Korean Pectobacterium strains
Whole-genome sequence analysis was conducted with seven Pectobacterium strains considered representatives of South Korean Pectobacterium (Table 1). Since JR1.1 was isolated from radish, a rare host, we selected two strains of Pca for comparison with the strain isolated from potato. To obtain a single chromosome, de novo assembly was performed with three programs for each strain. The best results were selected considering the low contig number, high BUSCO score, and longest sequence length with an average coverage of 300├Ś. The BUSCO score of the seven genomes ranged from 96.0% of MYP201603 to 98.0% of JR1.1. The genome sizes of the strains ranged from 4,724,337 bp observed with HNP201719 to 5,208,618 bp observed with HC, with a GC content ranging from 50.4% in HC to 52.3% in HNP201719 (Table 1, Fig. 1). The average number of CDSs of the seven strains varied, including 4,563 in JR1.1, BP201601.1, and HNP201719; 4,815 in JK2.1; and 5,161 in MYP201603, PZ1, and HC. The number of repeat regions ranged from 135 in HNP201719 to 226 in PZ1. A plasmid was identified in JK2.1 and MYP201603. Two plasmids showed a size of approximately 10 Mb and 120 CDSs. Li et al. (2018) reported that Ppa had the lowest average GC content, and Pbr had the highest average GC content among the six species. These findings are similar to the results of this study, but the GC content (52.3%) observed in HNP201719 was slightly higher than the 52.18% observed in other studies (Li et al., 2018, 2019). Complete whole-genome sequencing was performed with the genomes of seven strains, and it was expected to serve as an additional reference for whole-genome analysis of the Pectobacterium genus.
To evaluate conservation and variations among South Korean Pectobacterium strain collections, whole-genome sequences were aligned using the Mauve software. As this comparison was made at the species level, low-similarity regions were frequently observed among the genomes (Fig. 1). BP201601.1 showed a large inversion of local collinear blocks compared to JR1.1; however, the ANI value (%) was 98%, as observed with BP201601.1 and JR1.1 (Fig. 2A). The ANI values ranged from 88% to 96% at the species level of Pectobacterium in this study. Pve and Pod strains clustered more closely with Pca than with other strains. The Ppa strain, HC, showed the lowest ANI values compared to the other five species. PZ1 and NCPPB3395 considered as type strains of Ppv showed 99% identity in ANI value and 94% identity in the isDDH value. PZ1 was also negative for citrate utilization (Supplementary Table 2). Pan-genome phylogeny analysis was also performed with 13 reference strains for the accurate classification of seven isolated strains (Fig. 2B, Supplementary Table 1). BP201601.1 was more similar to WPP14 than to JR1.1, but both were classified as the same Pca species with an isDDH value of 70% (Li et al., 2019; Pasanen et al., 2020). Pasanen et al. (2020) reported 96.0-96.2% ANI identity and 66.1-68.4% isDDH value between Ppo and Ppv, and this report was similar with the result in this study. On comparing the utilization of four carbon sources, only Ppv isolates could not utilize citrate, among seven species of Pectobacterium including Ppo (Pasanen et al., 2020). The negative response of the PZ1 strain to citrate utilization was considered another piece of evidence supporting the re-classification of PZ1 from Ppo to Ppv (Supplementary Table 2). In this study, PZ1 was newly grouped with Ppv strain NCPPB 3395 and separated from Ppo type strain NIBIO 1006 upon examination of BIOLOG test, ANI value, isDDH value, and phylogenetic tree.
Cluster of orthologous group analysis of South Korean Pectobacterium collection
Cluster of orthologous group (COG) analysis was conducted with the seven Pectobacterium strains isolated in South Korea to compare the distribution of functional genes (Fig. 3). An average of 90.7% of the predicted CDSs was classified using the 25 COG categories, and approximately 29.3% were hypothetical proteins. No strain showed genes in five COGs including the general function prediction only group (R), the chromatin structure and dynamics group (B), the extracellular structures group (W), the nuclear structure group (Y), and the cytoskeleton group (Z); meanwhile, all strains had one gene of the RNA processing and modification group (A). The largest COG category in this analysis comprised the amino acid transport and metabolism group (E) with an average of 369 genes, followed by the carbohydrate transport and metabolism group (G) with an average of 331 genes, the inorganic ion transport and metabolism group (P) with an average of 318 genes, and the transcription group (K) with an average of 314 genes. Pca, JR1.1, and BP201601.1 showed similar distribution of functional genes. The number of genes showed a deviation of more than 10% among seven strains in the four COGs including the lipid transport and metabolism group (I); the replication, recombination, and repair group (L); the secondary metabolites biosynthesis, transport, and catabolism group (Q); and the intracellular trafficking, secretion, and vesicular transport group (U). The cell cycle control, cell division, chromosome partitioning group (D); the coenzyme transport and metabolism group (H); and the translation, ribosomal structure, and biogenesis group (J) showed a similar distribution of functional genes among the seven strains. MYP201603 had more functional genes in 11 groups. P. aroidearum L6 was different in three COGs groups including the defense mechanisms group (V), the cell motility group (N), and the signal transduction mechanisms group (T) (Xu et al., 2021). The Pba strain SX309 differed from HNP201719 in the N group and the T group (Li et al., 2019).
Comparison of enzymes distributed in the genomes with the analysis of CAZymes and PCWDEs
CAZyme analysis was conducted with seven strains to identify various enzymes in Pectobacterium. All strains showed similar distributions in the CAZymes class (Fig. 4). The average of glycoside hydrolases (GHs) was 44.5%, glycosyltransferases (GTs) was 26.4%, polysaccharide lyases (PLs) was 14.1%, carbohydrate esterases (CEs) was 8.0%, carbohydrate-binding modules was 5.4%, and auxiliary activity components (AAs) was 1.5% (Fig. 4A). HNP201719 and PZ1 showed the lowest total number of CAZymes, whereas HC demonstrated the highest number. The highest number of two CAZyme classes, namely, CEs and GTs, was identified in HC. HC and JK2.1 had the highest number of GHs. Known PCWDEs were identified in the genomic sequence of the isolated strains (Supplementary Table 3). This analysis focused on secreted PCWDEs with signal peptides, including pectin-degrading enzymes, cellulases, and proteases. The presence of pectin-degrading enzymes and cellulase was similar among five strains, JR1.1, BP201601.1, JK2.1, HNP201719, and MY201603. HC strains lacked at least one enzyme in the three categories of enzymes compared to other strains. JK2.1, HNP201719, and MYP201603 showed more proteases than JR1.1 and BP201601.1 of Pca.
He et al. (2021) conducted CAZyme analysis with Pca strain PC1, grouped in Par by Li et al. (2018). He et al. (2021) identified 144 CAZymes, including 57 GHs, 35 GTs, 26 CEs, 15 PLs, and 8 AAs. The number of AAs and CEs in PC1 was higher than 1-2 in AAs and 8-12 in CEs in this study. The number of PLs was conserved in the Pectobacterium species. Several enzymes with signal peptides in the CAZymes analysis were identified in PC-WDEs (Supplementary Table 3). These enzymes in the CAZyme category were considered an important tool for mechanisms underlying pathogenesis (He et al., 2021). Three main peptidase families, serine, metallo-, and cysteine super-families, were identified at more than 80% of secreted peptidase genes of 2,191 bacterial genomes, and the metallopeptidase family was more diverse than the serine-peptidase and cysteine-peptidase families (Nguyen et al., 2019). Figaj et al. (2019) referred to Pectobacterium as a phytopathogen with the best-characterized metalloproteases such as Prt1 and PrtW. In the extracellular protease of this study, four serine protease genes were identified along with eight metalloproteases, consistent with the results of Li et al. (2019).
Comparison of genetic variation in T1SS, T2SS, and T3SS among Pectobacterium strains
To understand the pathogenicity of Pectobacterium strains, we compared the gene clusters of T1SS, T2SS, and T3SS among the strains and identified three subtypes of T1SS and two copies of the lapBCE subtype in this collection (Fig. 5A). Pectobacterium strains had four T1SS gene clusters, except for the HC strain of Ppa. T1SSa was not detected in the HC group. The T2SS gene cluster typically comprises 15 structural genes linked to pel and pehX in Pectobacterium strains. T2SS was highly conserved in the genomes of the six strains, but HC did not show gspN and pel (Fig. 5B). Six Ppa strains, including type strains, also did not demonstrate T1SSa similar to HC, and three subsets have been identified in Pectobacterium species (Li et al., 2018). In other studies, P. aroidearum strain L6 showed 14 genes, and P. aroidearum strain PC1 demonstrated one gene (He et al., 2021; Xu et al., 2021). Further study on T1SS and protease secretion is required since Ppa strains, including HC with three T1SS, did not show protease activity in the medium (Jee et al., 2020). T2SS was identified to be highly conserved in this study, similar to other studies (Li et al., 2018, 2019). Xu et al. (2021) reported 23 genes with loss of gspS in the T2SS of strain L6. This conservation could be attributed to the importance of T2SS, which secretes extracellular PCWDEs used as the main weapon by Pectobacterium.
Three subsets of T3SS were also identified in the Pectobacterium strains (Fig. 6A). The gene cluster of T3SSa was conserved in five strains, namely JR1.1, BP201601.1, JK2.1, HNP201719, and MYP201603. All strains had T3SSb, which encodes flagellar components and was highly conserved. Except for the flagellar system, HC did not demonstrate T3SS, and PZ1 showed T3SSc, an alternative to T3SSa. Several SPI-1-related genes were identified in T3SSc. Fig. 6B shows a comparison of the effectors of T3SS. Effector genes of Pectobacterium, such as dspE, dspF, and hrpK, were identified in all strains except for HC and PZ1. Owing to the query coverage of 16%, hrpK of JK2.1 is not shown in Fig. 6B. The putative T3SS effector, srfC-like gene, was identified in JR1.1, HNP201719, and HC. PZ1 demonstrated cysteine protease gene yopJ and cysteine methyltransferase gene ospZ. The putative effector srfC has been observed from plant to animal bacterial pathogens (Pallen et al., 2005). DspE is known to induce plant cell death in Pectobacterium, but the role of two putative effectors, hrpK and srfC-like genes, remains unknown (Arizala and Arif, 2019). Loss of T3SSa was observed in six Ppa strains and the Ppo strain NCPPB3395, reclassified as Ppv (Li et al., 2018; Pasanen et al., 2020). Pasanen et al. (2020) identified SPI-1-like T3SS in four of the 18 strains of P. parvum. We identified T3SS genes and the effector, namely yopJ and ospZ, of human pathogens such as Salmonella, Shigella, Yersinia, and Escherichia in PZ1. sipDC is known to participate in the formation of the needle complex in SPI-1 T3SS and aids in pathogen internalization and secretion of effectors (Lou et al., 2019). The gene invJ controls the length of the needle segment, and invF regulates the outer proteins (Sops) of Salmonella. invB encodes a chaperone for SopA. The original function of the yopJ effector is to mediate immunosuppressive reactions, which was identified in Yersinia species. The nleE/ospZ effector was identified in enteropathogenic Escherichia spp., Shigella spp., and Salmonella spp. nleE/ospZ effector of PZ1 showed approximately 67% homology with that of Salmonella enterica, and yopJ effector in PZ1 showed 48% homology with that of Yersinia species in BLAST. YopJ family effectors are widely distributed in diverse bacteria and are homologous to the HopZ effector of Pseudomonas syringae (Ma et al., 2006).
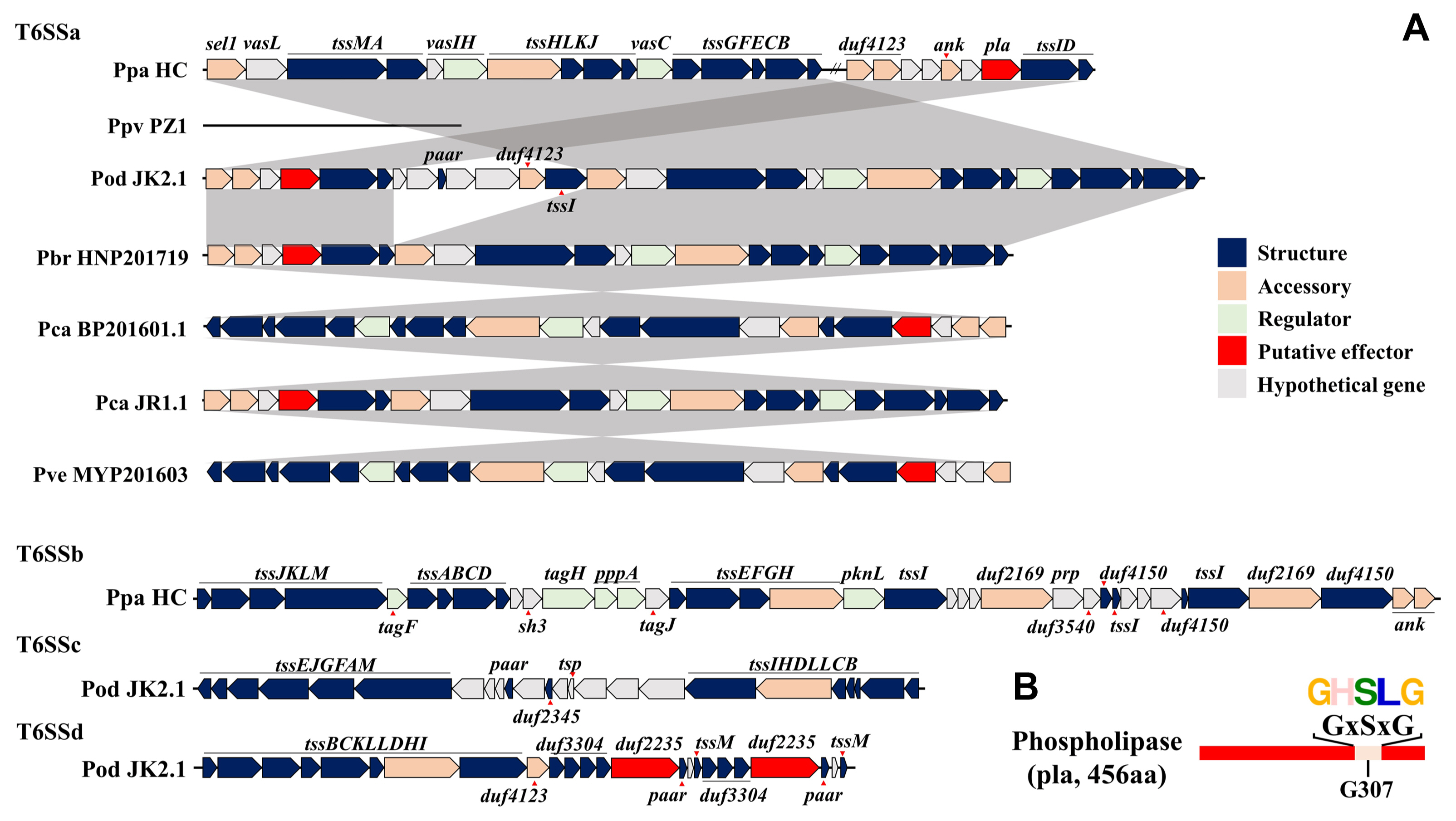
Comparison of T6SS and putative effector genes revealed in the genome of Pectobacterium strains
We identified 13 structural genes, six regulator genes, and five accessory genes in the T6SS gene cluster of seven Pectobacterium species (Fig. 7A). Structural genes included tssA-G, tssI-M, and paar. Regulator genes included vasC, vasH, tagF, tagH, serine/threonine-protein kinase (pknL), and serine/threonine phosphatase (pppa). Accessory genes comprised putative chaperone genes, including tssH, duf2169, and duf4123, and putative immune genes, including sel1-like repeat (sel1) and ankyrin repeat domain (ank). We identified T6SSa, a conserved subset, in four strains, JR1.1, BP201601.1, HNP201719, and MYP201603. We also observed the loss of T6SS in PZ1. HC showed a longer distance between the needle complex and other components of T6SSa and demonstrated a higher number of regulatory genes and accessory genes in T6SSb compared to other strains. JK2.1 showed three subsets of T6SS and an incomplete composition of structural genes in T6SSc and T6SSd. No tssK was observed in T6SSc, and tssA, tssE, tssF, and tssJ were absent in T6SSd. In the T6SSa cluster, the phospholipase A gene (pla) was identified between tssID and duf4123 for all strains. tssID represents valine-glycine repeat encoding gene (vgrG) and hemolysin-coregulated protein-encoding gene (hcp), known as the core components of the needle complex (Cascales and Cambillau, 2012) and effectors in T6SS of Pectobacterium species (Mattinen et al., 2007).
The DUF4123 protein family has been described as an effector-specific chaperone or adapter. As one of the conserved catalytic motifs of antibacterial phospholipase effectors (Russell et al., 2013), the GxSxG motif is located on G307A in the phospholipase A protein (Fig. 7B). This PLA showed a sequence length of 456 aa and was highly conserved among Pectobacterium strains. JK2.1 showed two DUF2235 domain-containing genes with the predicted protein of 805 aa and 813 aa in T6SSd. In JK2.1, this domain was classified as an alpha/beta hydrolase domain and demonstrated a GxSxG motif on G244A and G246A. T6SS is a bacterial machine used to inject an effector to manipulate the host and communicate or fight other bacteria. This system has 13 core components encoded within the same gene cluster (Bernal et al., 2018). T6SS resembles a phage tail and consists of four structures: a contractile sheath constituted by TssB and TssC, a baseplate structure constituted by TssA, TssE, TssF, TssG, and TssK, an integral membrane complex constituted by TssL, TssM, and TssJ, and the needle complex as an inner tube constituted by VgrG (TssI), Hcp (TssD), and PAAR (Bernal et al., 2018). A chaperone is required for the interaction or secretion of effector and VgrG/PAAR structures. Immune proteins are required for self-protection against effectors in attacking cells.
All strains showed T6SS except for PZ1 as Ppv, which was identified in P. parvum strain NCPPB3395. Many T6SS effectors show antibacterial activity, such as peptidoglycan amidase, peptidoglycan glycoside hydrolase, lipase/phospholipase, pore-forming protein, DNase, and NAD(P)+ glycohydrolase (Coulthurst, 2019). pla is located in the T6SS cluster of the genome of six South Korean Pectobacterium strains and shows the known motif of phospholipase A effector, type VI lipase effectors (Tle) 1-4. duf2235, identified in T6SSd, belongs to the Tle1 family, and is known to possess phospholipase A1 and phospholipase A2 activity (Jana and Salomon, 2019). The hcp genes are known along with three families as chaperones of effectors, including duf4123, duf1795, and duf2169, usually identified upstream of their cognate effectors (Jana and Salomon, 2019). This finding additionally supports the role of pla and duf2235 as effectors. To date, only Hcp and VgrG are considered effectors in Pectobacterium, and this study is the first report showing phospholipase as a putative effector of T6SS in Pectobacterium species.
Two to 14 hcp/vgrG/paar islands were identified for each Pectobacterium strain (Fig. 8). Single hcp varied from 0 to 10 for each strain, but a copy of single vgrG was observed in HC and JR1.1. A single paar was not identified in any of the strains. The hcp-vgrG set was identified with one or two copies in all strains, including PZ1, which did not demonstrate T6SS. P. wasabiae SCC3193 had 26 genes for hcp or vgrG, and P. wasabiae WPP163 showed 18 genes for hcp or vgrG (Nykyri et al., 2012). This is a higher number than that reported here, although Nykyri et al. (2012) did not report hcp/vgrG/paar islands. Several domains presumed to have specific functions were found in the hcp/vgrG/ paar islands of seven strains, including duf4123, duf2235, alpha/beta hydrolase, duf2931, virus-type replication-repair nuclease domain (vrr-nuc), and duf3396. duf4123 could be considered a putative chaperone, and duf2235 and alpha/ beta hydrolase could be considered as an effector with phospholipase activity. duf2931 was suggested as putative immunity proteins on downstream of Tle family effector in Aeromonas hydrophila (Ma et al., 2020). Wang et al. (2021) identified TseV-TsiV as a putative effector-immune protein pair, which showed a VRR-NUC domain and DUF3396. VRR-NUC and DUF3396 remain unknown in the Pectobacterium genus, and ours is the first report to demonstrate the presence of this pair in Pectobacterium.
Our results showed that seven representative strains were classified as six species, Pbr, Pca, Pod, Ppa, Ppv, and Pve, with the ANI, isDDH, and phylogenetic tree. The number of PCWDEs was 30-32 in all strains except for HC in which it was 26. Four subtypes of T1SS were identified in six strains. Three subsets were identified in T3SS, and T3SSb was conserved in seven strains. HC showed three subsets of T1SS with the loss of T1SSa and a subset of T3SSb. SPI-1 like T3SS structural gene and effectors were identified in T3SSc of PZ1. Two putative effectors including hrpK were identified in seven Pectobacterium strains; however, their function was unknown. In T6SS, JR1.1, BP201601.1, HNP201719, and MYP201603 demonstrated a subset of T6SSa, while PZ1 did not include any subset. HC had two subsets including T6SSa and T6SSb, and JK2.1 showed three subsets of T6SS. T6SSa cluster genes harbor a pla gene located among tssID and duf4123 genes in six Pectobacterium strains. Phospholipase is a representative effector of T6SS with a unique motif, GxSxG, identified in the pla of six Pectobacterium strains. Considering this, the pla gene is a putative effector of T6SS in Pectobacterium. Although Pasanen et al. (2020) referred to the neighboring lipase in T6SS of Ppo, the authors did not identify the lipase as an effector. Important domains were identified in the hcp/vgrG/paar islands, including the duf2235 gene and alph/beta hydrolase, which potentially has phospholipase activity in BP201601.1, HC, and MYP201603, and the vrr-nuc gene, which was referred to as a putative effector with nuclease activity in HC. This report identified the putative effectors with pla gene, duf2235, and vrr-nuc domains in Pectobacterium strains. Nevertheless, further studies are warranted to unravel their specific functions in Pectobacterium strains.





 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print