Comparative Analyses of Four Complete Genomes in Pseudomonas amygdali Revealed Differential Adaptation to Hostile Environments and Secretion Systems
Article information

Abstract
Pseudomonas amygdali is a hemibiotrophic phytopathogen that causes disease in woody and herbaceous plants. Complete genomes of four P. amygdali pathovars were comparatively analyzed to decipher the impact of genomic diversity on host colonization. The pan-genome indicated that 3,928 core genes are conserved among pathovars, while 504–1,009 are unique to specific pathovars. The unique genome contained many mobile elements and exhibited a functional distribution different from the core genome. Genes involved in O-antigen biosynthesis and antimicrobial peptide resistance were significantly enriched for adaptation to hostile environments. While the type III secretion system was distributed in the core genome, unique genomes revealed a different organization of secretion systems as follows: type I in pv. tabaci, type II in pv. japonicus, type IV in pv. morsprunorum, and type VI in pv. lachrymans. These findings provide genetic insight into the dynamic interactions of the bacteria with plant hosts.
As a hemibiotrophic phytopathogen, Pseudomonas amygdali is a gram-negative, motile, and rod-shaped bacterium. This bacterium is one of the P. syringae species complex including more than 60 pathovars. A genomic comparison study, which used a DNA-DNA hybridization, suggested nine genomospecies within the P. syringae species complex (Cardan et al., 1999). Genomospecies 2, the largest cluster, comprised five previously named Pseudomonas species and 26 pathovars of P. syringae, primarily belonging to woody and herbaceous phytopathogens (Cardan et al., 1999). Through this study, many pathovars initially assigned to P. syringae, such as morsprunorum, lachrymans, tabaci, mori, and sesami, were ascribed to P. amygdali.
Among P. amygdali pathovars, pv. morsprunorum, which has released the most genomic information from more than P. amygdali 20 strains, is a causal agent of bacterial canker in Prunus species. It was reported that up to 75% of sweet cherry trees were lost due to disease by these phytopathogens (Hulin et al., 2018). The pv. lachrymans, which contains 15 P. amygdali strains with genomic information, causes the bacterial angular leaf spot, resulting in a 30% to 50% reduction in cucumber yield across China (Meng et al., 2017). Another major group, pv. tabaci is responsible for tobacco wildfire disease, whose outbreaks have recently increased due to climate change (Taguchi and Ichinose, 2011). Given the wide host range and economic damage from various pathovars, an understanding of the dynamic interactions of P. amygdali with plant hosts is essential to protect agricultural crops. However, little research has comprehensively assessed the functional diversity among P. amygdali pathovars.
Comparative genome analysis is an efficient approach to determine the genetic diversity of strains in the same species and is fundamental for defining its pan-genome. It consists of three gene types: core (conserved genes), dispensable (shared by a subset of strains), and unique (strainspecific genes). The core genome is responsible for basic biology across the strains, whereas the unique genome allows strain-specific adaptation to the environment (Tettelin et al., 2008). The composition of the unique genome in phytopathogens is continuously remodeled according to dynamic interactions with plant hosts (Dudnik and Dudler, 2014). Phytopathogens will acquire countermeasure strategies through the unique genome to cope with host defense mechanisms. Several studies showed that comparative genome analysis is efficient in uncovering the pathogen-specific strategies, such as signal recognition, effector production, and biofilm formation (Bocsanczy et al., 2017; Hersemann et al., 2017).
Our study applied comparative genome analysis of P. amygdali pathovars for deciphering genetic determinants for host colonization. We focused on three P. amygdali pathogens, including pv. lachrymans NM002, pv. tabaci 6605, and pv. morsprunorum R15244, for which complete genomes have been reported. The complete genomes are better than partial genomes to detect the complete hereditary information in evolutionary history (Mardis et al., 2002). Lukjancenko and Ussery (2014) noticed that core genes in pan-genome analysis sharply dropped when considering partial genomes, which possessed the incomplete genetic information. The accuracy and order of all genes are necessary to infer genomic events with bacterial adaptations among closely related strains. We first investigated the phylogenomic distribution of the 66 P. amygdali strains consisting of 14 pathovars, which are available for partial (>scaffold assembly) or complete genomic information. From 712 concatenated genes derived by the Roary tool (https://sanger-pathogens.github.io/Roary/), a phylogenomic tree revealed distinct clades of 1–12 strains supported by the current pathovar structure (Fig. 1). The pv. lachrymans NM002, pv. tabaci 6605, and pv. morsprunorum R15244 were positioned in major clades, indicating that three pathogens are closely related to the other strains within each pathovar. Moreover, the pathogenicity of the three pathogens was experimentally verified (Hulin et al., 2018; Li et al., 2019; Taguchi et al., 2010).
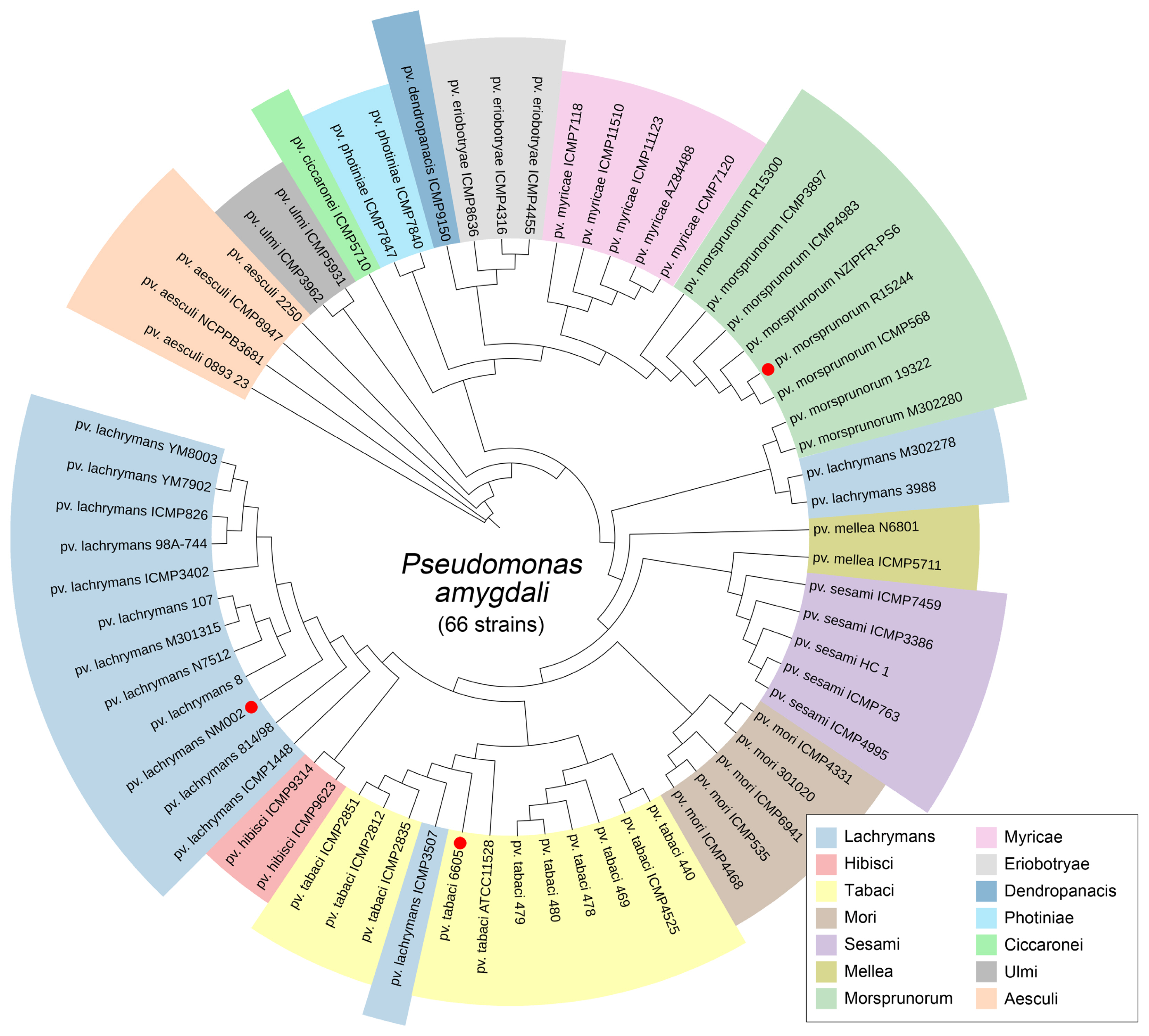
Phylogenomic distribution of Pseudomonas amygdali pathovars. Genome sequences of 66 P. amygdali strains were compared using the Roary tool with a 95% similarity cutoff and 10−5 E-value. A total of 712 genes were concatenated into one string to construct a maximum-likelihood tree using the RAxML tool. Each pathovars group was painted with the assigned color. The strains used for genome comparison in this study are highlighted in red circles.
We further generated a complete genome of P. amygdali HS1 isolated from infected leaves of Mallotus japonicus trees. HS1 strain causes leaf spot symptoms with clearedged circular or irregular shapes with discoloration in M. japonicus. M. japonicus, distributed mainly in East Asia, has an ecological importance for insects and herbivores interactions, and its extracts are used as cosmetic and medical additives (Satomi et al., 1994). Since 2014, diseases caused by this pathogen have been continuously observed in Jeolla and Jeju provinces in South Korea (Yun and Kim, 2021). A 16S rRNA-based phylogenetic tree derived from 100 P. amygdali strains indicated that HS1 strain is a clear separation between pv. morsprunorum and pv. tabaci as a new pathovar (Supplementary Fig. 1). Therefore, the increased availability by the complete genome of pv. japonicus HS1 will allow robust measurements of the pan-genome to cover functional distribution among P. amygdali pathovars.
The genomic features of the four P. amygdali pathovars used in comparative analysis are summarized in Table 1. All genomes showed a small deviation in size (5.93–6.29 Mb) and GC content (58.0–58.3%). Moreover, the numbers of rRNAs, tRNAs, and other RNAs were almost identical. The average nucleotide identity (ANI) is the most used bioinformatic approach to calculate the global similarity among genomes. Recent studies reported that ANI values >96% are the threshold for the same species (Zhang et al., 2016). Consistent with the genomic features, ANI values among P. amygdali pathovars were between 96.3% and 98.7%, of which pv. tabaci 6605 had the highest identity (98.7%), with pv. lachrymans NM002 (Fig. 2A). The pv. japonicus HS1 genome shared 96.3–96.4% ANI values with those of the other strains. Next, the pan-genome analysis was performed to determine the core, dispensable, and unique genomes (Fig. 2B). The Roary tool with a 95% similarity cutoff and 10−5 E-value was used to cluster the orthologous genes. The calculated pan-genome included 3,928 core genes, accounting for 71.4–79.1% of each genome. Besides the core genome, the pan-genome possessed unique genomes containing from 504 to 1,009 pathovarspecific genes.
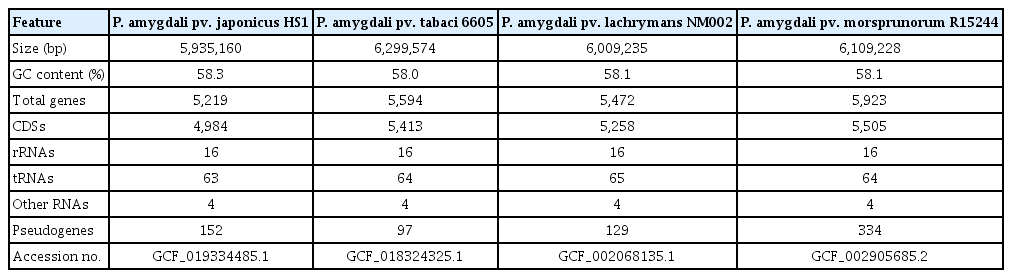
Genomic features of four Pseudomonas amygdali pathovars
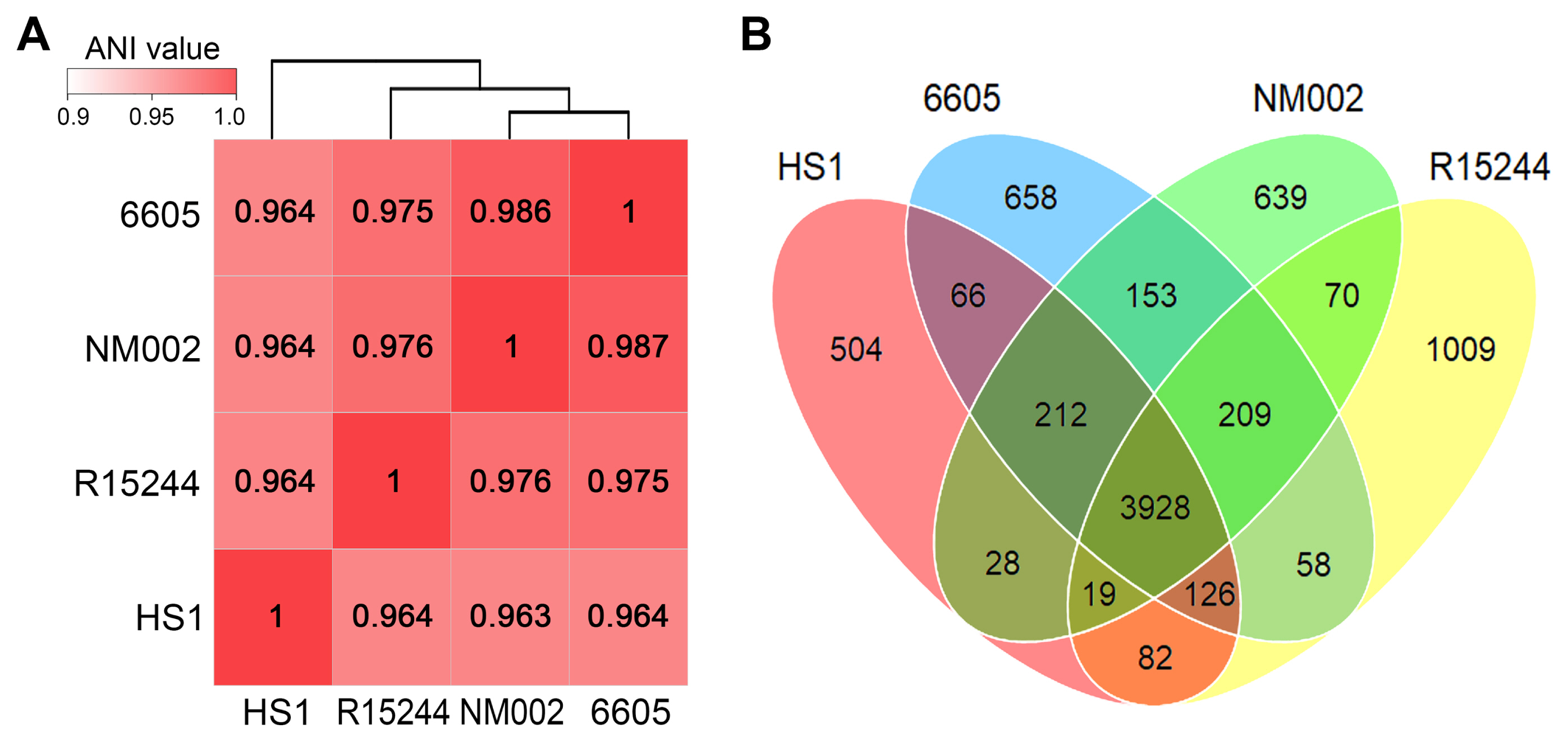
Genomic relationships between Pseudomonas amygdali pathovars. (A) The average nucleotide identity (ANI) values were calculated by PyANI script using BlastN alignment. The display range of the genomic similarity is given by the color bar. Strains were clustered via a hierarchical dendrogram based on similar values. (B) The distribution of pan-genome was determined using Roary. The Venn diagram shows the number of unique and shared orthologous genes between the genomes.
To estimate functional distribution across the pangenome components, the clusters of orthologous groups (COG) database was analyzed with the BlastP tool (E-value < 10−5 and >30% in both coverage and identity) (Fig. 3). The core genes were dominant with functions such as amino acid transport and metabolism (10.7%), signal transduction mechanisms (8.6%), transcription (8.1%), and translation, ribosomal structure, and biogenesis (6.0%). These functions are involved in the flow of genetic information as the central dogma, which demonstrate a conservation for basic functions within P. amygdali pathovars. By contrast, the unique genes exhibited a different distribution from that of the core genes. It was noted that, in pv. lachrymans NM002, about 45% of them belonged to the mobilome: prophages, transposons that primarily encode transposases and integrases. These components contribute to the increase in genetic diversity, which is responsible for the introduction of virulence factors (Oliveira et al., 2014). The replication, recombination, and repair function highly distributed in the unique genomes may support the expansion of genetic elements in phytopathogens. Moreover, the COG categories evidenced a heterogeneous functional pattern of the unique genomes of P. amygdali pathovars. For instance, unique genes of pv. japonicus HS1 showed a higher rank of cell wall/membrane/envelope biogenesis (8.9%) that serves to protect phytopathogens from hostile environments.

Distribution of clusters of orthologous groups (COG) functions among pan-genome components. Genome groups are presented on the x-axis and relative abundance of COG categories on the y-axis. Each COG category corresponds to a distinct color, as shown on the right.
To predict the metabolic capability of the unique genomes, they were annotated using the Kyoto Encyclopedia of Genes and Genomes database (https://www.genome.jp/kegg/). The enrichment analysis was performed using a hypergeometric test (P < 0.05). The degree of enrichment was calculated by the rich factor reflecting the ratio of unique genes to the total number of annotated genes in each pathway. Several pathways were significantly enriched in the unique genomes, although the genomes showed insufficient genetic composition (Fig. 4A). The O-antigen nucleotide sugar biosynthesis was uniquely enriched in pv. tabaci 6605. The O-antigen is the main component of the bacterial lipopolysaccharide and is responsible for diverse plant-pathogen interactions. Recent studies suggested that the composition and structure of O-antigen are crucial determinants of the virulence potential even within the bacterial strains (Lerouge and Vanderleyden, 2002). Modifying the O-antigen in phytopathogen Xylella fastidiosa, for example, reduces the perceptive ability of bacterium and enables altered effects of the host innate immune system (Rapicavoli et al., 2018b). The cationic antimicrobial peptide (AMP) resistance was the most enriched pathway in pv. japonicus HS1. AMPs are ubiquitous in eukaryotes and are important defense molecules of the innate immune system. Several pathogens have overcome the AMPs-mediated cell lysis through various mechanisms such as surface remodeling, expressing efflux pumps, and proteolytic degradation (Pandin et al., 2016; Schmidtchen et al., 2002). Pandin et al. (2016) reported that soft-rot phytopathogen Dickeya dadantii possesses homologs of the dlt genes that alter its membrane to resist AMPs. Taken together, these systemic functions belonging to each unique genome among P. amygdali pathovars may be involved in bacterial adaptations related to evasion of the host immune system.

Enriched Kyoto Encyclopedia of Genes and Genomes pathways in unique genomes. (A) Scatter plots indicate significantly enriched pathways in the unique genomes. Rich factor refers to the ratio of the number of unique genes to the total number of annotated genes in each pathway. The size of the dots is corresponding to the number of enriched genes, and its color marks statistical significance. (B) The type III secretion system was visualized based on the core genome of Pseudomonas amygdali pv. japonicus HS1. (C) The bacterial secretion modules were visualized based on the unique genome of each P. amygdali pathovar. Structural components of modules assigned to core and unique genes are highlighted in red text.
The pv. lachrymans NM002 and pv. morsprunorum R15244 unique genomes were highly enriched with genes for biofilm formation and bacterial secretion (Fig. 4A). Paradoxically, these pathways were also found in other two strains. Biofilm formation participates in all stages of the plant disease cycle, from inoculation to the establishment of infection. Phytopathogens produce various components to make biofilms for host colonization (Koczan et al., 2011; Yu et al., 1999). Thus, it was inferred that the unique genome drives biofilm diversity among P. amygdali pathovars according to the host environment. Indeed, the results showed several biofilm types, including biofilm formation in Escherichia coli, Vibrio cholerae, and Pseudomonas aeruginosa. In pv. morsprunorum R15244 unique genome, the enriched bacterial chemotaxis and two-component system pathways involved in biofilm biosynthesis also support our hypothesis (Fig. 4A).
While a type III secretion system (T3SS) was distributed in the core genome shared by all P. amygdali pathovars (Supplementary Table 1), the unique genomes contained different secretion systems. The T3SS is a conserved virulence factor in pathogens of both plants and animals. The pv. japonicus HS1 genome also constituted the T3SS consisting of 14 core genes (Fig. 4B). The T3SS directly injects type III secreted effectors (T3SEs) into host cytoplasm, allowing pathogens to subvert host immunity and manipulate cellular activities (Cunnac et al., 2009). Many studies revealed that the P. syringae species complex harbor the largest repertoire of T3SEs known to play a central role in virulence (Chang et al., 2005; Guttman et al., 2002). On the other hand, the evolutionary process of acquiring type I (T1SS), type II (T2SS), type IV (T4SS), or type VI (T6SS) secretion systems in P. amygdali pathovars seems to be faced with selective pressures imposed by each host.
The pv. tabaci 6605 presented the T1SS consisting of ATP-binding cassette transporter, membrane fusion proteins, and outer membrane factor (Fig. 4C). The T1SS allows secreting virulence proteins such as proteases, lipases, and toxins into the host extracellular environment. The phytotoxin syringomycin in P. syringae species, which causes cell lysis in plant hosts by altering the membrane potential, is a representative example secreted by T1SS (Bender et al., 1999). The pv. tabaci 6605 synthesizes a monocyclic beta-lactam tabtoxin, known as the wildfire toxin (Kapchina et al., 2014). It spreads radially from infection sites causing tissue yellowing and chlorosis. Although its secretory mechanism is unknown, tabtoxin production may be associated with the T1SS. It was found that the outer membrane gene Pta6605_RS24120 in the T1SS is shared with the efflux pump gene in beta-lactam resistance (Supplementary Fig. 2). Therefore, pv. tabaci 6605 may have employed an additional module specialized to both secrete and self-protect virulence factors.
The unique genes of pv. japonicus HS1 were assigned to the inner membrane complex of the T2SS (Fig. 4C). In phytopathogenic Proteobacteria, most T2SS-secreted proteins are specialized to degrade the plant cell wall. Goo et al. (2010) revealed that many cell wall-degrading enzymes, including lipase and proteases, are secreted through the T2SS in phytopathogen Burkholderia glumae. It was reported that mutating the T2SS gene xpsE in X. fastidiosa confers a nonpathogenic phenotype reminiscent of a mutant lacking a cell wall-degrading enzyme (Rapicavoli et al., 2018a). The unique genome of pv. japonicus HS1 may also be related to the secretion of enzymes to break down the cell wall of M. japonicus. The inner membrane and pilus complexes of the T2SS and T4SS share interesting similarities from the structural and functional standpoint. They secrete similar toxins (e.g., cholera toxin in the T2SS and pertussis toxin in the T4SS) and use a similar DNA uptake mechanism (Lawley et al., 2003). Compared with other systems, the T4SS can transport DNA–protein complexes across cell membranes. Our data indicated that the unique genome of pv. morsprunorum R15244 encodes the integral components of the T4SS from the ATPases to pilus proteins (Fig. 4C).
The T6SS is a recently characterized system that injects effectors into the host cell. It was revealed that nine unique genes of pv. lachrymans NM002 were annotated in the T6SS (Fig. 4C). The T6SS is required not only for virulence mechanisms but also for the adaptation to environmental signals. Low pH is an effective host defense strategy that inhibits phytopathogen colonization. Yuan et al. (2008) demonstrated that the T6SS of phytopathogen Agrobacterium tumefaciens, the causal agent of crown gall disease, is induced by acidic conditions (pH 5.5) of plant tissues. Furthermore, mutating a core T6SS gene in A. tumefaciens reduced virulence on potato tuber (Wu et al., 2008). Considering the acidic environment at pH 5.1 in the cucumber host, the T6SS will be used as a unique countermeasure for the acidic adaptation of pv. lachrymans NM002.
Historically, a lack of understanding of the fundamental physiology of the Pseudomonas species had led to the idea of a single species complex or limited classification. Our study provides valuable insights to the genomic diversity among P. amygdali pathovars. Highly conserved regions with >96% ANI values were observed in all complete genome sequences. Nonetheless, the pan-genome map showed that unique genomes of each pathovar indicated different functionalities from the core genome. These genomic properties contribute to pivotal mechanisms for the successful colonization of plant tissues, including the O-antigen biosynthesis and AMP resistance. Furthermore, although many unique genes were common in the bacterial secretion systems, different modules revealed their necessity for dynamic interactions with hosts. The work presented here demonstrates a suitable platform for functional genomics of P. amygdali species: a valuable model towards establishing strategies against these phytopathogens.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2019R1A2C2006779) and by a grant from the Nakdonggang National Institute of Biological Resources (NNIBR), funded by the Ministry of Environment(MOE) of the Republic of Korea (NNIBR202202108).
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Electronic Supplementary Material
Supplementary materials are available at The Plant Pathology Journal website (http://www.ppjonline.org/).