In accordance with the international trend to integrate the domestic and overseas markets, the Korean fruit industry has recently become quite globalized. However, the simultaneous increase in the cultivation of fruit trees and reduction in pollinating insects has resulted in a sharp increase in artificial pollination. Over 90% of the artificial pollen used on fruit trees is currently imported from other countries (Australia, China, Japan, and countries in North and South America and Europe) (Desvignes, 1985; Hernández and Flores, 1992; Osaki et al., 1999; Shamloul et al., 1994). Therefore, there is an increased possibility of inflow of foreign pathogens into Korea through imported pollen and an increased risk of transmission of those plant pathogens.
To date, 39 plant viruses (most belonging to the Alphacryptovirus, Ilarvirus, Nepovirus, or Potyvirus genera) have been confirmed to be pollen-transmitted; additional six plant viruses are tentatively considered to be pollen-transmitted (Card et al., 2007). In addition, five viroids have been reported to be transmitted through pollen (Card et al., 2007). This is important because the quality or quantity of pollen produced by viruses or virus-infected plants may be reduced compared to that of healthy plants. Many viruses and viroids are transmitted through pollen; however, despite their effect on the quantity and quality of pollen, the viral population of imported pollen has not been reported to date. Therefore, knowing the inflow of viruses and viroids and their route of transmission in the imported fruit pollen are important for preventing the inflow of quarantined viral pathogens into the country.
In general, plant viruses are detected and identified using serological and molecular biological methods (Zhang et al., 2017). Enzyme-linked immunosorbent assay (ELISA) (serological method) can diagnose several samples inexpensively and quickly, but its sensitivity is limited (Torrance and Jones, 1981). Therefore, a more specific and sensitive detection of plant viruses is done by molecular diagnosis (Maliogka et al., 2018). The detection of viruses by molecular-biology methods such as polymerase chain reaction, quantitative real time polymerase chain reaction (qPCR), and loop mediated isothermal amplification is more sensitive than ELISA and offers multiple detection possibilities (López et al., 2003; Lu et al., 2018; Malandraki et al., 2017; Osman et al., 2015). However, molecular biological methods can only detect known viruses, and they have limited ability to detect recently discovered viruses and to discover novel viruses. These problems can be overcome using next generation sequencing (NGS) technology, wherein the sequence data of all putative viral agents present in the sample can be generated without the prior knowledge of the viral genome (Radford et al., 2012).
High-throughput sequencing is a highly efficient bioinformatics tool with the ability to process a larger number of samples simultaneously, thus reducing sequencing costs (Osaki et al., 1999). Additionally, the ease with which samples and libraries can be prepared is another key advantage (Huang et al., 2019). High-throughput screening can be used to simultaneously detect RNA viruses, DNA viruses, and viroids in a variety of plant samples containing very low titers. Plant viruses can be easily diagnosed and identified using this innovative technology (Qian et al., 2014).
NGS has been used in plant virology since 2009 (Adams et al., 2009; Al Rwahnih et al., 2009; Kreuze et al., 2009). This advent of NGS revolutionized plant virology as a powerful method for identifying plant viruses without any a priori knowledge (Massart et al., 2017). NGS has been used in studies including, but not limited to, the discovery of novel viruses, detection and identification of known pathogens, analysis of genomic diversity and evolution, and pathogen epidemiology (Hadidi et al., 2016). In addition, the technology has provided new insights through the identification of the causes of plant viral disease in crops, understanding the abundance and diversity of viruses, screening specific viruses in suspect plants, and detecting asymptomatic viruses (Akinyemi et al., 2016; Roossinck et al., 2015). NGS technology is progressively reaching the field of plant virus diagnostics, and is also making an impact in the field of quarantine (Martin et al., 2016; Massart et al., 2014). So far, several plant viral metagenomic studies have identified novel viruses and viroids in several crop species, including apple (Jakovljevic et al., 2017; Wright et al., 2020), grapevine (Al Rwahnih et al., 2011; Czotter et al., 2018; Jo et al., 2018c), citrus (Matsumura et al., 2017), lilies (Jo and Cho, 2018; Li et al., 2018), vanilla (Grisoni et al., 2017), barley (Jo et al., 2018a), pepper (Jo et al., 2017), and peach (Jo et al., 2018b).
In this study, comprehensive bioinformatics analyses of plant viruses in imported pear and kiwifruit pollen were performed using RNA-sequencing (RNA-seq) to investigate viruses and viroids transmitted through imported pollen. In addition, we revealed the diversity of viruses and viroids infecting pear and kiwifruit pollen.
Materials and Methods
Sample preparation
For artificial pollination, 20 g of pear and kiwifruit pollen imported from China in 2019 were purchased from the commercial market, three pieces each. These pollen samples were pooled, grounded in liquid nitrogen, and stored at −80°C.
RNA extraction and library preparation for RNA-seq
Pollen RNA was extracted using the IQeasy Plus Plant RNA Extraction Mini Kit (iNtRON, Seongnam, Korea) according to the manufacturer’s instructions. The quantity and quality of purified RNA samples were measured and evaluated using the Biochrom Libra S70 Double Beam Spectrophotometer (Biochrom Ltd., Cambridge, UK). The quality of RNA was measured with the BioAnalyzer 2100 (Agilent Technologies, Palo Alto, CA, USA) and RNA 6000 nano chip. Ribosomal RNA was removed from total RNA using a Ribo-zero rRNA removal kit for plants (Illumina, San Diego, CA, USA), and RNA was purified and recovered using RNA Clean XP. The TruSeq stranded total RNA library prep kit (Illumina) was used to construct a cDNA library. The size, concentration and quality of the generated library were confirmed using the Agilent D5000 ScreenTape system (Agilent Technologies) and LightCycle qPCR; and the flow cell in which the cluster was formed through the library clustering process was sequenced in Illumina Hiseq 4000, generating 151 bp paired-end reads. For the generated raw data, the base quality score was confirmed with the Fast QC program, and low-quality data (≤ Q20) and the adapter at the read end were removed using the Trim galore program before analysis.
Transcriptome assembly and virus identification
The raw data obtained from the library was subjected to de novo transcriptome assembly using gsAssembler v2.8 (Roche Diagnostics, Branford, CT, USA) using the default parameters (Wylie et al., 2014). From the contig obtained through the de novo transcriptome assembly, the contig blasted for e-value < 1e−10 for the mitochondria and chloroplast, and other host sequences of 200 bp or less in length was removed. To identify virus sequences in the library, the assembled contigs in the transcriptome were compared to NCBI viral reference database (https://www.ncbi.nlm.nih.gov/genome/viruses/) (Morgulis et al., 2008) using BLASTN, which is faster and more reliable for virus identification than other sequence similarity programs, and a cut-off E-value of 1e−5. Only virus-associated contigs were obtained for further pollen virome analyses.
Viral sequence mapping and genome assembly
To generate partial or complete viral genomic sequences, the pollen transcriptomes were first blasted against complete reference sequences of viruses. Contigs were selected based on the highest similarity to genomes of viruses in GenBank. For sequence alignment to the reference viral genome, virus-like reads were mapped to each reference viral genome using Geneious Prime 2020.2 (Kearse et al., 2012) and consensus sequences for each mapping file were generated based on a threshold of 95% identity.
Phylogenetic analyses of identified viruses
To analyze molecular genetic relationships for identified viruses, the complete or partial genome sequences of the coat protein (CP) of each virus were used in this study. For analysis of the CP gene sequence for each virus, different isolates were aligned using ClustalW in BioEdit 7.2 (Thompson et al., 2003). Phylogenetic analysis was performed using MEGA 7 (Tamura et al., 2011), and the maximum-likelihood algorithm was used to estimate the number of nucleotide substitutions per site in the Neighbor Junction (Rivarez et al., 2021) or Kimura 2 parameter method, then bootstrapped with 1,000 replications. The isolates were presented with NCBI accession numbers in the phylogenetic trees.
Confirmation of presence of identified virus by reverse transcription polymerase chain reaction
To confirm the presence of identified viruses by RNA-seq in pollen samples, reverse transcription polymerase chain reaction (RT-PCR) was performed using each virus-specific primer set to amplify genomic regions corresponding to the CP genes. Each of the 20 μl reactions included 0.4 μM forward primer, 0.4 μM reverse primer, 10 μl 2× SuPrimeScript RT Premix (Genet Bio, Daejeon, Korea), 2 μl template, and 6 μl DEPC-treated water. Amplification was performed under the following conditions: a reverse transcription step of 30 min at 50°C; initial denaturation at 95°C for 5 min; 35 cycles of 95°C for 30 s, 56°C for 30 s, and 72°C for 1 min; and a final extension of 5 min at 72°C. The RT-PCR products were analyzed through electrophoresis on 1.2% (w/v) agarose gel in 0.5× TBE buffer with RedSafe nucleic acid staining (iNtRON), and visualized under UV light. The size of the resulting RT-PCR product was confirmed by comparison with a 100-bp DNA ladder (iNtRON). In addition, the amplicons were cloned into the pGEM-T Easy Vector (Promega, Madison, WI, USA), and sequences were confirmed by Sanger sequencing.
Data availability
The raw dataset generated in the present study will be available upon publication in the NCBI Sequence Read Archive (SRA) repository under accession numbers SRR12614248 and SRR12614749. The viral genome sequences obtained from this study were also deposited in GenBank with their individual accession numbers.
Results
Sample collection and library generation
Transcriptome data were analyzed by RNA-seq for the identification of viruses in pear and kiwifruit pollen. Total RNA was isolated from pear and kiwifruit pollen samples imported from China, and two different libraries were prepared. Transcriptome data were obtained using paired-end sequencing from two different libraries (pear and kiwifruit) by HiSeq 4000, and the raw data of the sequenced reads by RNA-seq were deposited in an SRA database with their respective accession numbers (SRR12614248 and SRR12614749) (Table 1). The obtained raw data size was 7.2 Gb from pear pollen and 6.15 Gb from kiwifruit pollen, respectively. The number of obtained reads were 47,858,250 from pear pollen and 10,783,664 from kiwifruit pollen. The number of contigs, 82,285 from pear pollen and 271,201 from kiwifruit pollen, were obtained through data trimming and de novo transcriptome assembly (Table 1).
Identification of viruses from pollens
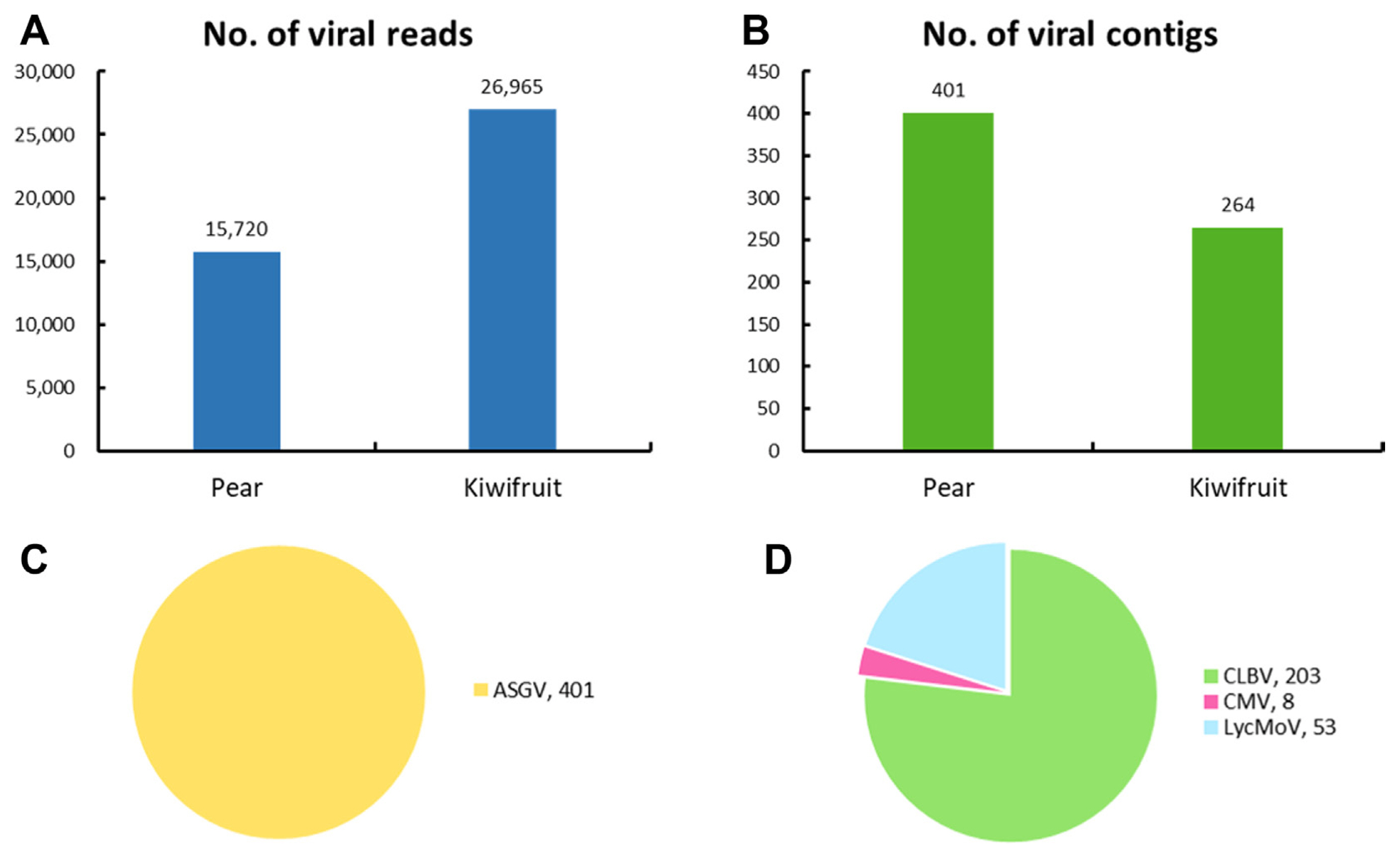
To identify virus-associated reads and contigs, the raw reads and assembled contigs were utilized and a BLASTN search against a virus reference genome database from NCBI was done. The number of virus-associated reads was 15,720 in pear pollen, while the number of virus-associated reads was 26,965 in kiwifruit pollen (Fig. 1A). Additionally, we identified a total of 665 virus-associated contigs (pear: 401 and kiwifruit: 264) in both types of pollen (Fig. 1B). The number of virus-associated contigs was 401 (apple stem grooving virus, ASGV) in pear pollen, while the number of virus-associated contigs was 203 (citrus leaf blotch virus, CLBV), 8 (cucumber mosaic virus, CMV), and 53 (lychnis mottle virus, LycMoV) in kiwifruit pollen (Fig. 1C and D).
Viral genome assembly
Virus-associated reads were mapped to each reference viral genome to obtain complete or near-complete genomes of viruses isolated from pear and kiwifruit pollen. ASGV isolated from pear pollen was mapped to 99.9% (Fig. 2A), while three different viruses isolated from kiwifruit pollen were mapped to 93.4 to 100% (Fig. 2B-D). In addition, the virus that obtained a 100% complete genome by read mapping was CLBV in kiwifruit pollen. The assembled contigs identified in each pollen were confirmed through a BLAST search, and as a result, ASGV was identified from pear pollen, and CLBV, CMV, and LycMoV were identified from kiwifruit pollen. The sequences of the CP of each virus were deposited in GenBank (Table 2).
Phylogenetic analyses of identified viruses
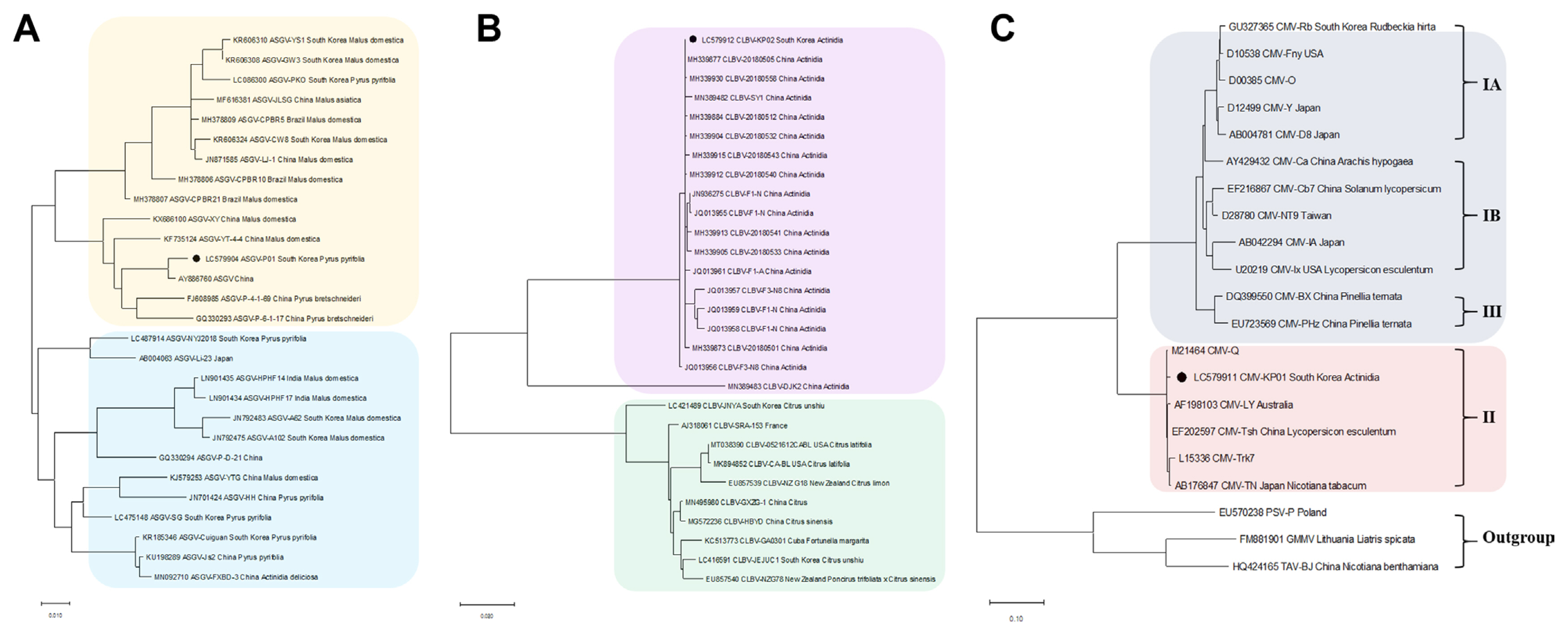
Phylogenetic analyses were conducted to determine the evolutionary relationship of the viruses identified from pear and kiwifruit pollen using RNA-seq. We retrieved CP sequences of 28 ASGV isolates, 29 CLBV isolates, and 21 CMV isolates including the identified isolates from this study from the NCBI database for pear and kiwifruit pollen (Fig. 3).
A phylogenetic analysis of the CP sequences of the ASGV-P01 (LC579904) identified from pear pollen, as well as other isolates from different countries, showed two separated clades; and the isolate was closely related to the ASGV isolate from China (AY886760, Pyrus serotine Reld) (Fig. 3A). For CLBV, the phylogenetic tree of CLBV showed two separate clades; with the CLBV-KP02 (LC579912) identified from kiwifruit pollen being closely related to CLBV-20180512 (MH339884, Actinidia sp.), CLBV-20180505 (MH339877, Actinidia sp.), and CLBV-20180558 (MH339930, Actinidia sp.) from China (Fig. 3B). Phylogenetic analysis of 21 CMV isolates revealed that CMV-KP01 identified in this study clustered with the American isolate (M21464) from subgroup II (Fig. 3C). In the case of LycMoV, no phylogenetic analysis was performed due to the lack of available genome sequences of LycMoV deposited in the GenBank.
Validation of the identified viruses using RT-PCR
To confirm the presence of viruses identified in pear and kiwifruit pollens by RNA-seq, RT-PCR was performed using specific primers for the detection of these viruses. Virus-specific primers were designed based on the CP regions of identified viruses (Table 3). The results of RT-PCR were similar to those of RNA-seq (Fig. 4). Infection of ASGV in pear pollen was confirmed by RT-PCR (Fig. 4A). In addition, infections of CLBV, CMV, and LycMoV were validated using RT-PCR (Fig. 4B).
Discussion
Metatranscriptomics using high-throughput sequencing is a powerful and straightforward genome analysis technique for revealing the composition and abundance of viral communities, also referred to as the virome, in various environmental conditions, host species, and geographical distributions (Osaki et al., 1999; Rivarez et al., 2021). Due to the ease of identification of known or unknown viral pathogens from extremely large amounts of sequence data, RNA-seq is considered as an ideal technique for understanding the complexity of virus genomes and viral mutation in a certain environment. Thus, it has been introduced into more plant virome studies and diagnostic purposes (Villamor et al., 2019). Virome studies on fruit trees using RNA-seq have been extensively employed in the past few years (Jo et al., 2018a, 2020; Kim et al., 2022). However, few studies have been performed on the virome in fruit pollen samples (Jonghe et al., 2018). In this study, we used a metatranscriptomic pipeline using RNA-seq to define viruses infecting pear and kiwifruit pollen imported from China. RNA-seq revealed that the assembled virus-associated reads nearly completed assembly corresponding to target virus genomes, including ASGV, CLBV, CMV, and LycMoV. Only ASGV, one of the most common viruses infecting pear trees worldwide, was identified from pear pollen; and CLBV, CMV, and LycMoV, which are already reported in Korea, were identified from kiwifruit pollen, indicating that this study increases knowledge of viral diversity, also referred as virosphere, in fruit pollen.
From a biosecurity perspective, fruit pollen is a potential source of various viral fruit tree pathogens. Pollen is a valuable source of germplasm for breeding purposes in plants. In addition, pollination is a process in the reproduction of most spermatophytes and requires pollen and ovule for fertilization to occur (Card et al., 2007). Some viruses that have evolved mechanisms to utilize the plant reproductive process can be transmitted through pollen (Hull, 2002; Mink, 1993). Due to the increasing high risk of pollen-transmitted viral pathogens during the international trade of agricultural commodities worldwide, the International Plant Protection Convention set phytosanitary measures in accordance with the International Standard for Phytosanitary Measures No. 02 (1995) ‘Guidelines for Pest Risk Analysis’.
When the viruses are transmitted by pollen, the infection of plants through fertilized flowers is called horizontal transmission (Hull, 2013). In general, this horizontal transmission of viruses by pollen is caused by movement of the virus from the infected embryo to the maternal tissues (Card et al., 2007). Most viruses infecting fruit trees through pollen belong to Alphacryptovirus, Ilarvirus, Nepovirus, or Potyvirus (Card et al., 2007); however, the identified viruses in this study belong to genus Capillovirus (ASGV), Citrivirus (CLBV), Cucumovirus (CMV), and tentative new species in the family Secoviridae (LycMoV). Thus, further horizontal transmission assays are required to test whether these viruses are transmitted naturally through pollen via cross-pollination using fruit woody plants. Metatranscriptomic analyses do not provide information on the biological activity of identified viruses; thus, their viability should be assessed further. Our results also raise the possibility that contamination occurred during the collection process due to the nature of fruit pollen. This present study did not involve back inoculation (based on Koch’s postulates) in each healthy fruit tree for viruses identified in pollen, since identified viruses are generally transmitted through grafting and seeds, not by mechanical inoculation. Notably, fruit pollen may harbor pathogenicity and function as a vehicle for viral spread. ASGV is generally seed and mechanically transmitted, and no natural vectors have been reported (Kim et al., 2021). However, a recent study revealed that Nicotiana benthamina plants were infected with ASGV through pollination with ASGV-infected pollen grains derived from Malus pumila, indicating that pollen grains possibly served as a vector for horizontal pollen transmission of ASGV (Isogai et al., 2022). To date, 13 plant viruses, including CLBV and CMV, have been proven to naturally infect kiwifruit plants (Blouin et al., 2013). In the case of CLBV, CLBV is transmitted by grafting and seed in citrus. CMV belongs to the family Bromoviridae, which has a broad host range (with more than 1,000 host plant species in over 100 families); thus, Actinidia species as a host is not a new finding for CMV. CMV is generally transmitted by several types of aphids, seeds, and mechanical infections. LycMoV is a tentative new species in the family Secoviridae and was first reported in Lychnis cognate in South Korea (Yoo et al., 2015). Even though biological features of LycMoV has not been elucidated to date, it can be transmitted by mechanical means and seeds (Shaffer et al., 2019).
Isolates of ASGV, CLBV, and CMV identified from this study were clustered with Chinese isolates based on phylogenetic analyses, indicating that these viruses have a tight genetic association with those virus isolates in China. In addition, the viruses were of mixed infection with various variants in pollen samples, which presents the complexity for recombination, genetic diversity, and evolution. This mixed infection will possibly contribute to genetic exchanges, and thus producing new hybrid variants or novel viruses (through genetic recombination or reassortment), eventually leading to the expansion of the virosphere (Nabi et al., 2022).
In the current study, we identified ASGV, CLBV, CMV, and LycMoV from imported pear and kiwifruit pollen using RNA-seq, respectively, expanding the knowledge of the complexity of viral communities in fruit pollen. However, the pathogenesis mechanisms of these viruses through pollen transmission into pear and kiwifruit trees is still up for debate. With the increase of commercial trade of fruit pollen worldwide, further studies on fruit pollen virome are urgently needed, allowing the prevention of the inflow of high-risk viruses transmitted through pollen to other countries.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print