The Phytophthora genus comprises about 120 pathogens causing late blight disease to major vegetable crops such as pepper, tomato, and potato. Of those, 34 Phytophthora species have been identified as major plant pathogens in Korea between 1900 and 2000 (Hyun and Choi, 2014). The oomycete of P. infestans is causing one of the most devastating diseases in potato worldwide, including Korea (Kim et al., 2014). The disease is very infectious and can spread throughout the field within one week, leading to severe yield losses (Fry, 2008). This requires timely application of proper disease control measures at the early incidence stage to minimize crop infestation. The rapid evolution of P. infestans led to the prevalence of the virulent A2 mating type. The resistance of new P. infestans genotypes to fungicides has rendered difficult disease control challenging (Arora et al., 2014). To develop more effective genetic and chemical disease control methods, the population structure of P. infestans, the genetics of both mating types, virulence factors, and fungicide response of each strain have been extensively studied (Cooke and Lees, 2004; Göre et al., 2019; Rekad et al., 2017). Application of molecular techniques such as mitochondrial (mt) DNA sequence haplotyping, restriction fragment length polymorphisms (RFLPs), amplified fragment length polymorphisms, and simple sequence repeat (SSR) accelerated our understanding of the genetics and pathology of P. infestans (Drenth et al., 1993, May and Ristaino, 2004, Van der Lee et al., 1997). Carter et al. (1990) used RFLP technique to identify mt haplotypes in relation to mating types. Griffith and Shaw (1998) modified the RFLP method by adopting polymerase chain reaction (PCR) followed by restriction enzyme digestion. By using multiple restriction enzyme combinations, Martin and Tooley (2004) were able to design a PCR-RFLP method applicable to intra- and interspecific mtDNA haplotyping, which they used to calculate genetic relationship within 152 isolates of 31 Phytophthora species. This approach has been widely implemented for haplotype identification of regional P. infestans strains in South America (Cárdenas et al., 2011), Africa (Shimelash et al., 2016), and Asia (Guo et al., 2010). Mitochondrial haplotypes of P. infestans were used to track the origin, migration, and genetic diversity of global Phytophthora species (Gómez-Alpizar et al., 2007, Goss et al., 2014; May and Ristaino, 2004; Peters et al., 2014). Later, sequencing of the whole mt genome identified two hypervariable regions (HVRs): HVRi with 1,885 bp no insertion/deletion between trnY and rns genes, which were used to differentiate type I and II; and HVRii, carrying one, two, or three copies of 36 bp variable number of tandem repeats (VNTRs) between cox2 and trnW genes, which were used to further classify mtDNA as R1, R2, and R3 haplotypes. By this new PCR approach developed by Marin and Tooley (2004), the previously described four mtDNA haplotypes Ia, Ib, IIa, and IIb were reclassified into five types, IR1, IR2, IR3, IIR2, and IIR3 (Yang et al., 2013).
Genetic and pathological evidence supports the intercontinental migration of P. infestans further to Asia and displacement of previously dominant haplotypes by new types. Koh et al. (1994) reported the presence of US-1 (mating type A1) and JP-1 (mating type A2) in East Asian countries. The US-1 is found worldwide, including in China, Japan, Korea, and the Philippines, whereas JP-1 is reported only for Korea and Japan. Gotoh et al. (2005) extended their survey to wider Asian regions and discovered 14 new P. infestans haplotypes. Two genotypes, JP-1 and KR-1, were identified in Korea, with the latter being specific to Korea. Zhang et al. (2006) conducted extensive analysis to reveal genetic diversity of P. infestans populations in South Korea. They collected 367 isolates from major potato production locations between 2002 and 2004 and identified four genotypes, JP-1, US-17, US-14, and KR-1, based on allozyme patterns and mating types; JP-1 was the most frequent type across all locations and years. At least four genotypes, KR_1_A1, KR_2_A2, US-11, and SIB-1, were identified in Korea by 12 SSR markers (Choi et al., 2020).
However, a few reports exist on complete mt genome sequences of different P. infestans isolates (Kubo et al., 2011; Lassiter et al., 2015; Patil et al., 2017) and there are no reports on Korean P. infestans isolates. In the present study, we reported whole mt genome sequences of four Korean P. infestans isolates and haplotypes, and conducted a comparative analysis using previously reported mt genome sequences to delineate genomic diversity of this species in Korea.
Materials and Methods
Phytophthora infestans isolates and DNA preparation
Four P. infestans isolates, Kpi16-9, Kpi15-14, Kpi15-3, Kpi15-10, representing four different genotypes revealed by 12 SSR markers (Choi et al., 2020) were selected for mtDNA sequence analysis (Table 1). Single isolates were collected after cultivation on V8 fungal culture medium (8.5 g agar, β-sitosterol 0.025 g, 450 ml distilled water, 50 ml V-8 juice, 0.75 g calcium carbonate). The isolates were grown in pea liquid medium (120 g of frozen peas, 1 liter of distilled water) at 18°C with shaking at 160 rpm for 2 weeks. Mycelia were harvested from pea liquid media and washed with distilled water to remove the liquid medium. Mycelia were dried by blotting with sterilized paper towels and freeze-dried in 1.5 ml tubes before storing at −80°C. Total genomic DNA of P. infestans were extractedfrom 50 mg freeze-dried mycelia using a Fungi/Yeast Genomic DNA isolation kit (Norgen Corp., Thorold, ON, Canada) following manufacturer’s instructions.
Mitochondrial genome assembly
The next-generation sequencing (NGS) libraries were constructed using an Illumina Paired-End DNA library kit (Illumina, San Diego, CA, USA) with insert size of ~470 bp. The paired-end (PE) reads were sequenced in sets of approximately 8.0 Gbp using an Illumina HiSeq 4000 platform by Macrogen (Seoul, Korea). The overall de novo assembly method was followed by dnaLCW methods (Cho et al., 2015). The low-quality reads (less than Phred score of 20) were filtered using quality_trim script (CLC Assembly Cell package, version 4.2.1, Qiagen, Aarhus, Denmark). Then, high-quality PE reads were implemented in the de novo assembly by clc_novo_assemble tool with a distance between the forward start and reverse end reads controlled from 300 to 700 bp, minimum length fraction of read overlap of 0.5, and sequence similarity of 0.8 (CLC Assembly Cell package, version 4.2.1, Qiagen). The mt putative contigs were selected by nucmer program in MUMmer package (http://mummer.sourceforge.net/) and compared with P. infestans (AY898627) mt genome sequence as a reference. The ordering of selected contigs was based on the reference genome. Mapping of the contigs was confirmed by clc_ref_assemble tool in CLC Assembly Cell package (Qiagen). Mapping parameters were the same as those used for de novo assembly.
Mitochondrial genome haplotyping
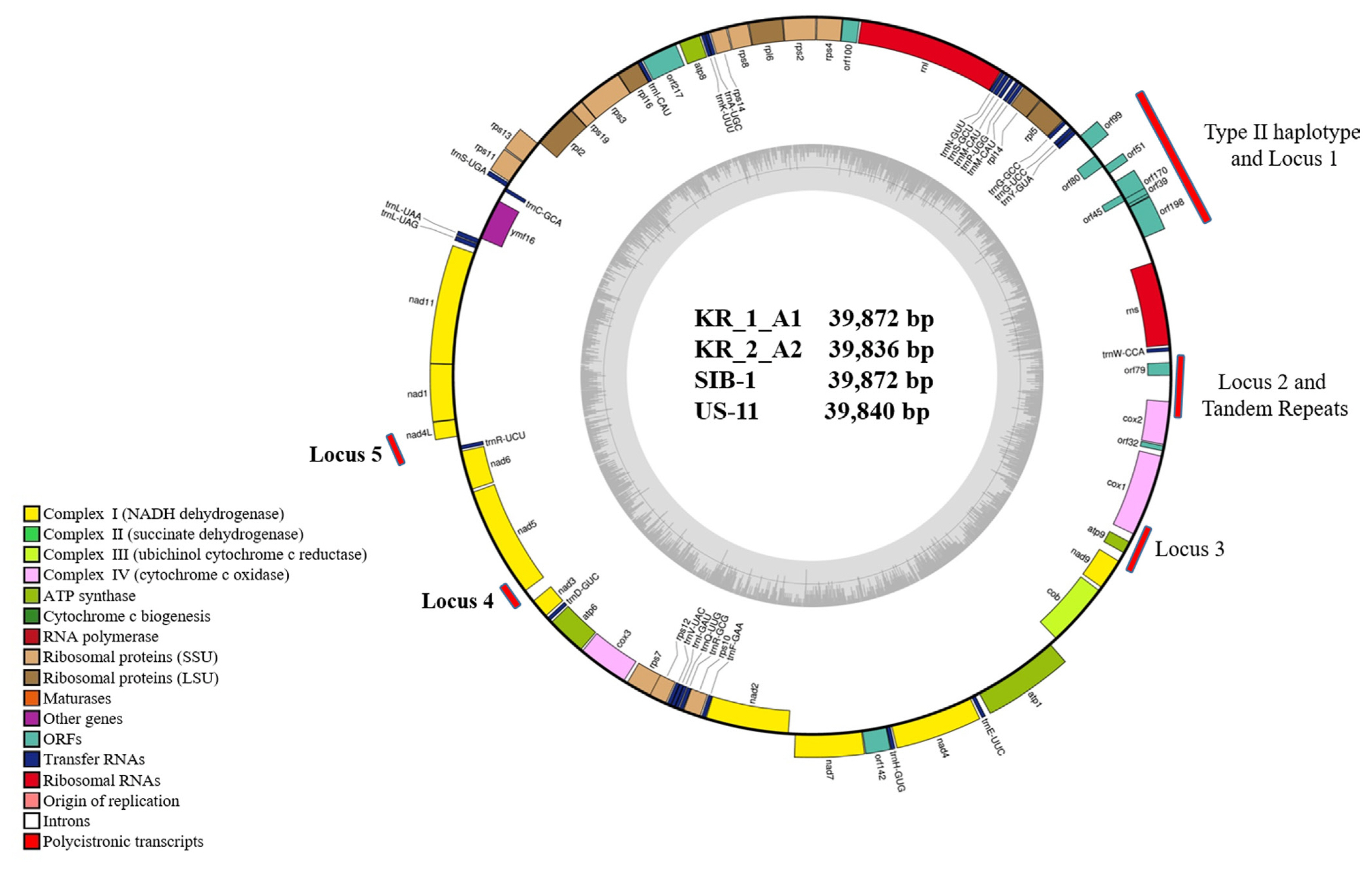
The mtDNA sequences of four Korean P. infestans isolates were analyzed to EcoRI and MspI restriction sites within the target regions (Carter et al., 1990; Griffith and Shaw, 1998) and grouped into Ia, Ib, IIa, or IIb haplotypes based on the sequences of predicted PCR-RFLP haplotypes by in silico. In addition to the HVRs carrying the 2 kb insertion at HVRi and 36 bp VNTR at HVRii between cox and orf79 genes (Yang et al., 2013) were also predicted to the haplotypes of mtDNA sequences. Five new haplotype loci developed by Martin et al. (2019) were analyzed (Fig. 1). In silico mt genome sequence analysis for haplotyping was conducted by CLC Main Workbench (version 20.0, Qiagen).
Minimal spanning network and phylogenetic analysis
Phylogenetic analysis of mt genome sequences of 12 Phytophthora species (Supplementary Table 1) from Martin et al. (2019) were conducted using maximum likelihood (ML) and maximum parsimony (MP) methods. The best fitting substitution model was K2 + G + I, selected based on the Bayesian information criterion and Akaike information criterion with correction (AICc) in MEGA X (Kumar et al., 2018). Node support was estimated using 1,000 bootstrap replicates in MEGA X (Felsenstein, 1985). The MP analysis was conducted in MEGA X with the subtree-pruning-regrafting algorithm of search level-1. The initial trees were obtained by random addition of sequences (10 replicates).
Results and Discussion
Complete mitochondrial genome sequences
Mitochondrial genome sequences of the four Korean P. infestans isolates were determined by NGS (Table 2). Mapping of the raw data onto the reference mt genome (NC_029397) revealed the average genome coverage of 570, 394, 241, and 389× in KR_1_A1, US-11, KR_2_A2, and SIB-1 P. infestans isolates, respectively. All the mt genomes were similar in size and gene content. The mt genome length of KR_1_A1 and SIB-1 (both 39,872 bp) was similar to that of US-11 (39,840 bp) and KR_2_A2 (39,836 bp) (Fig. 1). The KR_1_A1 and SIB-1 mt genome sequences were same in length. Each mt genome contained 61 genes, which included 2 RNA-encoding genes, 16 ribosomal protein genes, 25 transfer RNA and 17 genes encoding electron transport and ATP synthesis (NADH dehydrogenase, cytochrome b, c, ATP synthetase), 11 open reading frames of unknown function, and one import-related protein gene, tatC (Table 3). All the genomes were with the same gene order and G + C content of 22.2%. Coding sequences accounted for about 91.2% of the mt genome (Supplementary Table 2). None of the predicted genes contained introns, and ATG and TAA were the start and stop codons for all the genes except nadh11, which had TGA as stop codon. Korkmaz et al. (2014) reported that among the three stop codons (TAA, TAG, and TAG), TAA is the most common in bacteria. Similarly, TAA was the most common stop codon in P. infestans mtDNA. Seven orf genes, orf99, orf80, orf51, orf171, orf45, orf39, and orf198, were identified in the 2 kb insertion found in type IIa haplotype previously reported by Avila-Adame et al. (2006).
Comparative analysis among four complete mitochondrial genome sequences of Korean P. infestans isolates
General characteristics of the complete mt genome sequences of KR_1_A1, KR_2_A2, US-11, and SIB-1 are presented in Table 2. The results showed a high level of similarity in the overall genome composition of the four Korean isolates. There were no differences in total complementary DNA sequences (CDS) (91.2%), G + C content (91.2%), total tRNA bases (1,882), and total tRNA length (75.3) in the four isolates. Moreover, all isolates showed similar CDS bases (36,348) and total repeats (1,738), except for isolate US-11 mt genome sequences (total CDS bases of 36,291 and 1,838 total repeats) (Supplementary Table 2). In the type IIb haplotype, orf178 gene was 60 bp shorter than orf198, which was 597 bp long. Orf45 and orf39 were nested in the orf170 gene (Table 4). These results indicate that gene content and their order in the mt genome were highly conserved among the four Korean P. infestans isolates.
Mitochondrial genome haplotyping
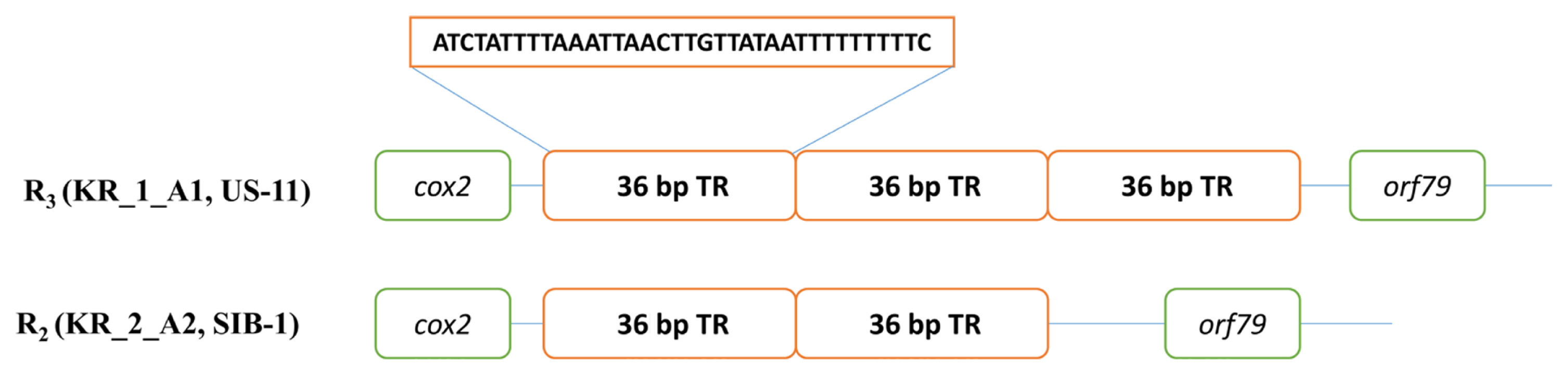
The sequence-based methods implemented in the present study identified seven mt haplotypes (Supplementary Data 1). The most polymorphic locus was the 1.1 kb rns to cox2 region (locus 2) with five haplotypes. There were seven single-nucleotide polymorphisms (SNPs; five conserved across type I and II haplotypes), 10 InDels (four 1 bp, one 2 bp, one 3 bp, one 4 bp, two 6 bp, and one 24 bp), and a variable number of the 36 bp repeat (one, two, three, or four copies located between orf79 and cox2) (Fig. 2). The next most polymorphic locus was nad6-nad4L (locus 5), the 0.5 kb region present in seven haplotypes, which comprised two, single-base InDels and four SNPs (one conserved between type I and II haplotypes). The remaining three loci rpl5-rns (locus 1), cox1-nad9 (locus3), and nad3-nad5 (locus 4) were three different haplotypes. The analysis of the five multilocus haplotypes generated a total of seven P. infestans multilocus mt haplotypes. Haplotype classification as followed by Martin et al. (2019) for example, haplogroup I-2 would be I-4-8-4-1-5, with locus 1 assigned to haplotype 4, locus 2 to haplotype 8 etc., while all the haplotypes belonged to haplotype IIa and IIb. Haplogroup IIa was the most prevalent, with 11 haplotypes among the five isolates (US-13, EU_8_A1, KR_1_A1, KR_2_A2, and SIB-1) compared with IIb, which had five haplotypes within the two isolates (US-11 and US-6). In haplotype IIb alone a US-11 isolate dominated in the group II haplotype.
Phylogenic analysis
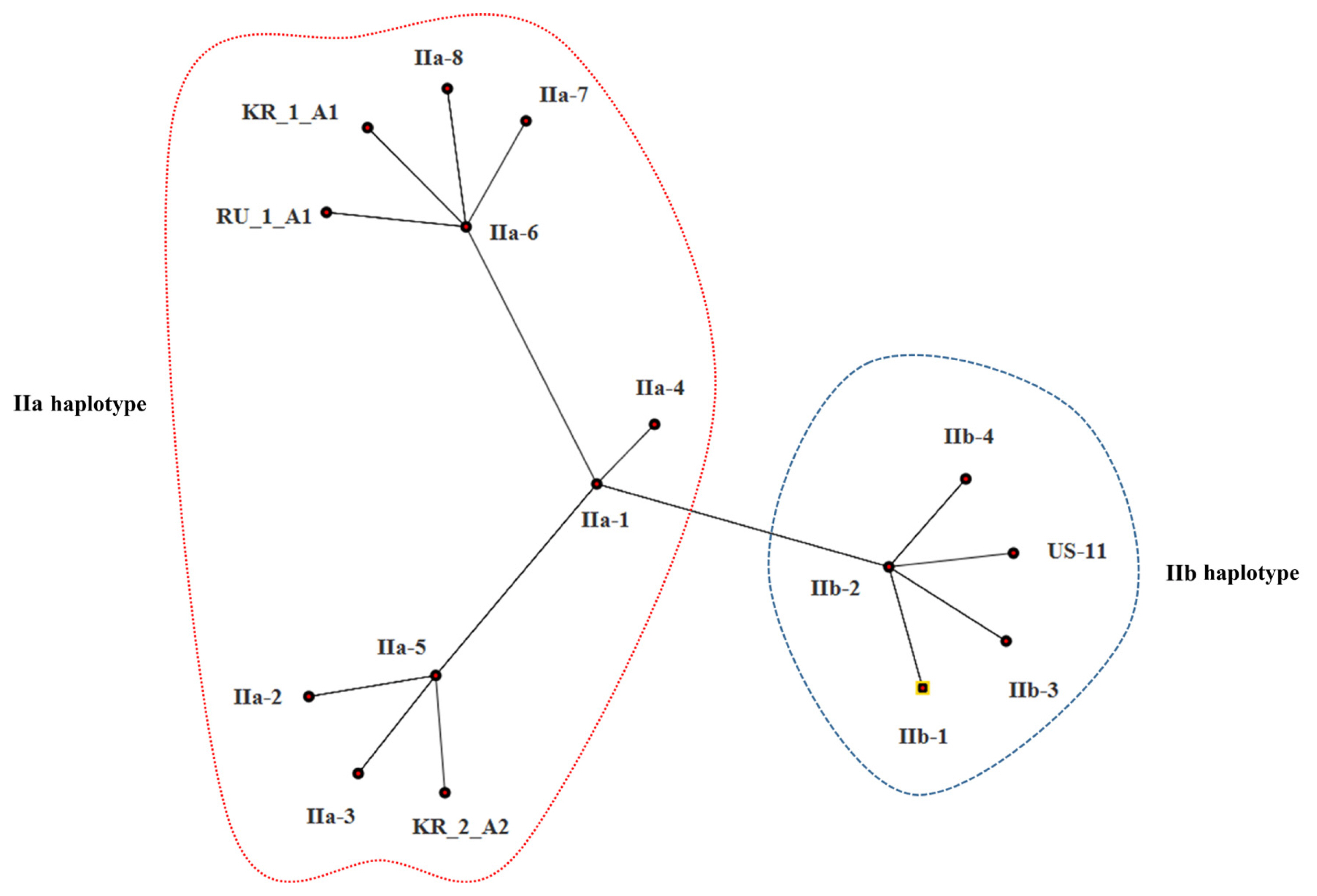
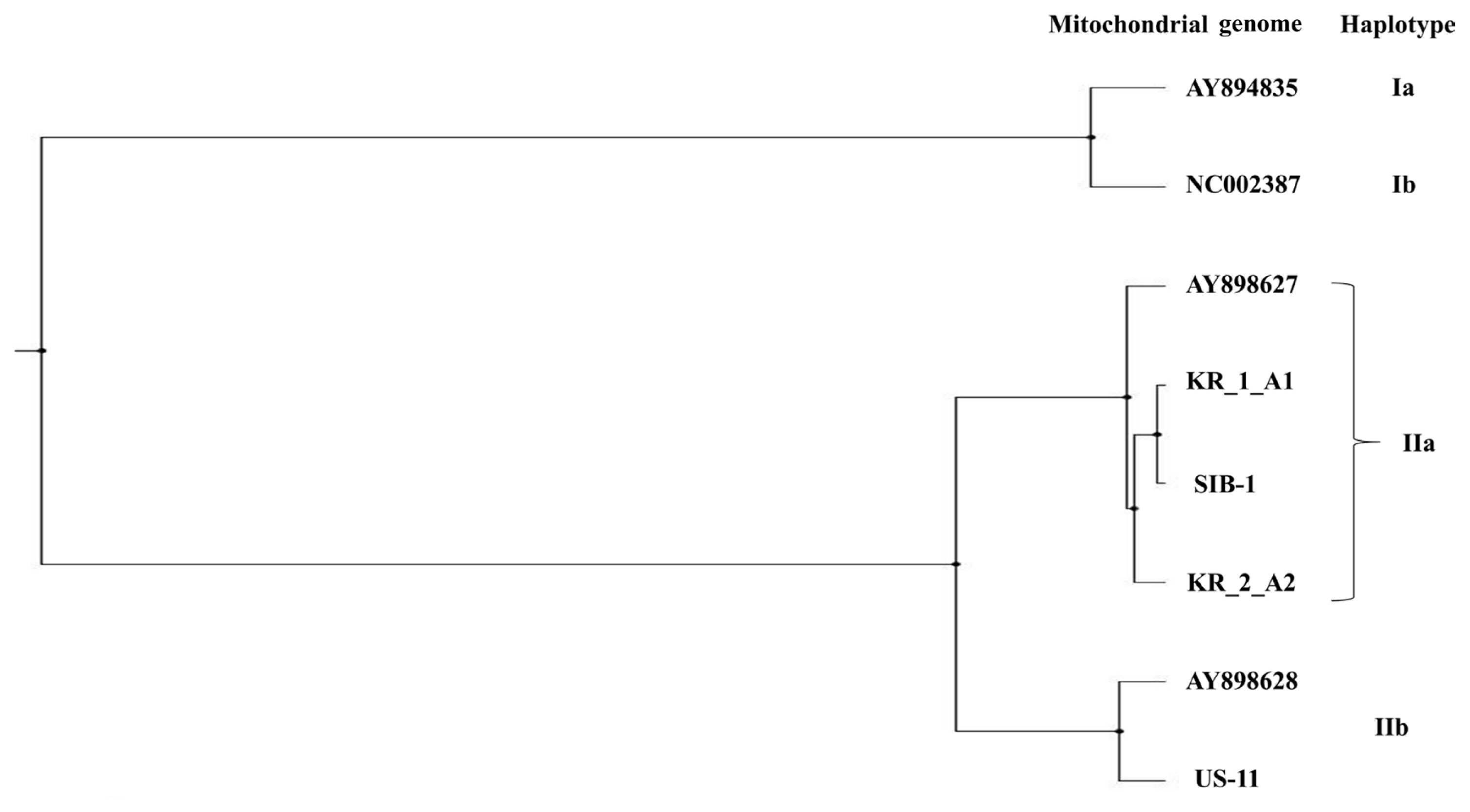
The estimates of evolutionary divergence between sequences are shown in Table 5. The sequence divergence between Korean and Russian isolates was small compared with the divergence between Korean and US isolates. Both MP and ML analysis resolved Korean isolates (KR_1_A1 and KR_2_A1) more divergent from the US isolate (US-11) than from the Russian isolate (SIB-1); of the two Korean isolates, KR_1_A1 was more closely related to the Russian isolate (SIB-1) (Fig. 3). The haplotype network analysis clearly identified two groups, IIa and IIb haplotypes (Fig. 4), within which Korean isolates and the Russian isolates were clearly divergent from the US isolates. KR_1_A1 and SIB-1 were branched from IIa-6 haplotype, as part of IIa haplotype. The MP analysis placed the Korean isolates with Russian isolates and distant to the US isolate. Within the Korean isolates, KR_1_A1 isolate formed a clade with SIB-1 (Fig. 5).
Migration of P. infestans in Korea
The potato was introduced to Korea in 1824 either from Japan in the south or from China in the north, with the latter being the more likely route (Cho et al., 2003). Akino et al. (2014) reported that Japanese populations of P. infestans might have been introduced to Japan from other countries such as Korea or China. SIB-1 is widely spread in northern China and northwest Russia (Siberia region), where it is a predominant strain (Elansky et al., 2001; Guo et al., 2010). This suggests that SIB-1 was introduced to Korea from the north, China or Russia, through contaminated potato seeds and then settled and spread throughout Korea. Considering the 100% match of mt genome sequences between SIB-1 and KR_1_A1, the Korean specific genotype likely originated from SIB-1. KR_2_A2, also found in Korea, might have been introduced from other countries or by changes in mating type that were triggered by exposure to fungicides (Goodwin and Drenth, 1997).

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement1
Supplement1 Print
Print