A Field Deployable Real-Time Loop-Mediated Isothermal Amplification Targeting Five Copy nrdB Gene for the Detection of ‘Candidatus Liberibacter asiaticus’ in Citrus
Article information

Abstract
Huanglongbing (HLB) is one of the most destructive diseases in citrus, which imperils the sustainability of citriculture worldwide. The presumed causal agent of HLB, ‘Candidatus Liberibacter asiaticus’ (CLas) is a non-culturable phloem-limited α-proteobacterium transmitted by Asian citrus psyllids (ACP, Diaphorina citri Kuwayama). A widely adopted method for HLB diagnosis is based on quantitative real-time polymerase chain reaction (qPCR). Although HLB diagnostic qPCR provides high sensitivity and good reproducibility, it is limited by time-consuming DNA preparation from plant tissue or ACP and the requirement of proper lab instruments including a thermal cycler to conduct qPCR. In an attempt to develop a quick assay that can be deployed in the field for CLas detection, we developed a real-time loop-mediated isothermal amplification (rt-LAMP) assay by targeting the CLas five copy nrdB gene. The rt-LAMP assay using various plant sample types and psyllids successfully detected the nrdB target as low as ~2.6 Log10 copies. Although the rt-LAMP assay was less sensitive than laboratory-based qPCR (detection limit ~10 copies), the data obtained with citrus leaf and bark and ACP showed that the rt-LAMP assay has >96% CLas detection rate, compared to that of laboratory-based qPCR. However, the CLas detection rate in fibrous roots was significantly decreased compared to qPCR due to low CLas titer in some root DNA sample. We also demonstrated that the rt-LAMP assay can be used with a crude leaf DNA extract which is fully deployable in the field for quick and reliable HLB screening.
Huanglongbing (HLB), also known as citrus greening, is one of the most destructive citrus diseases that imperils the sustainability of citriculture (Bové, 2006; Gottwald, 2010). HLB is associated with three Gram-negative phloem-limited α-proteobacteria species, ‘Candidatus Liberibacter asiaticus’ (CLas), ‘Ca. L. africanus’ (CLaf), and ‘Ca. L. americanus’ (CLam) (Bové, 2006; Jagoueix et al., 1994; Planet et al., 1995; Texeira et al., 2005). In the USA, HLB caused by CLas was first reported in Florida in 2005 (Halbert, 2005) and in California and Texas in 2012 (Kumagai et al., 2013; Kunta et al., 2012). CLas infection results in anatomical aberration of phloem which includes necrosis, starch accumulation and distortion of the cambial tissue (Wang et al., 2017). In addition, infected trees develop typical HLB symptoms in the tree canopy such as blotchy mottle, corky vein and lopsided fruit (Dala-Paula et al., 2019; McCollum and Baldwin, 2016). The disease diagnosis at early infection stage is difficult due to a lengthy latency period (da Graça et al., 2016; Gottwald et al., 2007). Therefore, it is likely that the presence of HLB in the field remains unnoticed leading to asymptomatic spread of the disease (Lee et al., 2015). As there is no cure for HLB, disease management practices, such as pesticide spray for insect vector control and foliar application of nutrients, have been put in place in an attempt to lessen the damage caused by HLB (Baldwin et al., 2012; Dala-Paula et al., 2019; Masaoka et al., 2011; Stansly et al., 2010; Zheng et al., 2018). Implementation of these methods is informed by HLB screening tools.
There have been many methods developed for HLB diagnosis, some of which include electron microscopy (Bové, 2006), enzyme-linked immunosorbent assays (Lu et al., 2013), conventional polymerase chain reaction (PCR) (Nageswara-Rao et al., 2013; Teixeira et al., 2005) and quantitative real-time PCR (qPCR) (Kim and Wang, 2009; Li et al., 2006; Morgan et al., 2012; Park et al., 2018; Wang et al., 2006; Zheng et al., 2016). Among these techniques, the qPCR-based methods are the most widely adopted method for HLB diagnosis. Although qPCR provides high sensitivity and reproducibility, it is time-consuming to obtain a final test result since the DNA template preparation and qPCR need to be conducted in the lab equipped with a thermal cycler and other required instruments.
Since 1990s, many different isothermal DNA amplification methods have been developed which eliminated the requirement of a thermocycler for DNA amplification (Kunta et al., 2021; Qi et al., 2018; Zanoli and Spoto, 2012; Zhao et al., 2015). Among these novel isothermal techniques, loop-mediated isothermal amplification (LAMP) (Notomi et al., 2000) and recombinase polymerase amplification (RPA) (Piepenburg et al., 2006) have been well adopted for the detection of pathogens due to the ease of technical accessibility and the excellent amplification specificity and efficiency (Daher et al., 2016; Misawa et al., 2007). Recently, the number of reports of isothermal assays for CLas detection has been gradually increased, all of which targeted different genetic loci of CLas genome. For example, CLas prophage region (Choi et al., 2018; Keremane et al., 2015), CLIBASIA_05175 encoding a hypothetical protein (Rigano et al., 2014) and nusG-rplKAJL-rpoB gene cluster (Okuda et al., 2005) were targeted for CLas detection by LAMP. For RPA assay, CLas 16s rDNA (Ghosh et al., 2018) and nrdB (Rattner et al., 2022) were targeted for CLas detection. Due to the variation of the target copy number in CLas genome, the detection limit of these isothermal assays could vary depending on the target genetic locus. CLas has five copies of nrdB gene which could be a good target gene for the improvement of CLas detection (Zheng et al., 2016). Rattner et al. (2022) recently reported an RPA assay for CLas detection by targeting CLas nrdB gene, which was coupled with a lateral flow detection system for the detection of target molecules amplified by RPA.
While both LAMP and RPA eliminated the multiple thermal cycling steps for target DNA amplification, LAMP and RPA are based on two different platforms. Unlike RPA that uses recombination proteins for the target DNA amplification (Piepenburg et al., 2006), LAMP uses DNA polymerases with strand displacement activity for the DNA amplification using 4 to 6 primers that recognize 6 to 8 distinct regions in the target sequence, resulting in greatly improved specificity compared to other isothermal amplification methods (Notomi et al., 2000; Tomita et al., 2008). This study developed a real-time LAMP assay (rt-LAMP) by targeting CLas five copy nrdB gene without the need of post-sample processing for CLas detection. The efficacy of the rt-LAMP assay for CLas detection was evaluated using various tissue samples that included citrus leaf, bark, root and Asian citrus psyllids (ACP). In addition, the study confirmed that the rt-LAMP assay can be conducted on a portable device, which verified the applicability of the rt-LAMP assay for CLas detection in the field.
Materials and Methods
Plant DNA and crude extract preparation
A total of 158 plant tissue samples (67 leaf samples with HLB or HLB-like symptoms), 58 fibrous roots, and 33 bark) were collected from grapefruit trees in an orchard located in Edinburg, Texas. Furthermore, 45 psyllids were collected across the Lower Rio Grande Valley (LRGV) of Texas. In addition, 14 plant tissue samples (7 leaf and 7 roots) were collected from healthy grapefruit trees maintained in the greenhouse to use as a negative control, which were propagated by Texas citrus budwood certification program.
Leaf and bark DNA fractions were prepared from collected samples using BioSprint 96 DNA Plant Kit (Qiagen, Germantown, MD, USA). Two hundred milligrams of finely chopped midrib and bark samples were collected in 2 ml screw-cap tubes (Thomas Scientific, Swedesboro, NJ, USA) containing two 1/4” ceramic spheres (MP Biomedicals, Solon, OH, USA) and a small amount of Garnet Matrix A (MP Biomedicals) for efficient tissue disruption. To each tube, 700 μl of RLT buffer was added, and the samples were homogenized in a Mini beadbeater 96 (Biospec Products, Bartlesville, OK, USA) for 4 min. All subsequent procedures for DNA extraction were conducted following the manufacturer’s instructions. Citrus leaf crude extracts were prepared in a sample mesh bag (Agdia, Elkhart, IN, USA) containing 100 mg of finely sliced midribs excised from three leaves and 500 μl of AMP1 extraction buffer (Agdia). The plant tissue in the sample mesh bag was crushed with a solid object such as pestle or pen from which the crude extract was collected in a microfuge tube for the downstream assay.
Before processing root tissue for DNA extraction, the collected fibrous root samples were kept in a paper bag for 24 h at room temperature (~25°C) to facilitate the removal of soil attached to the root samples (Park et al., 2018). After removing excess soil from the fibrous root samples by gently tapping the sample with fingertips, the root samples were finely sliced with a razor blade. The root DNA fraction was prepared using Qiagen DNeasy Plant Pro Kit (Qiagen) following the manufacturer’s instructions.
The DNA fraction was extracted from individual ACP adults using Qiagen DNeasy Blood and Tissue Kit (Qiagen). Psyllids were placed in a 2 ml screw-cap tube containing 360 μl phosphate-buffered saline, one 1/4” ceramic spheres and a small amount of Garnet Matrix A, which was then homogenized in a Mini beadbeater 96 for 2 min. The sample tubes were centrifuged for 10 min at 16,000 ×g, from which 180 μl of supernatant was transferred into a new microfuge tube. After adding 20 μl of proteinase K and 200 μl of buffer AL into the tube, the psyllid DNA fraction was prepared following the manufacturer’s protocol.
rt-LAMP primer design based on CLas nrdB gene
A full-length nucleotide sequence of nrdB gene of CLas strain A4 (GenBank ID: CP010804) was used to design primers for rt-LAMP assay for CLas detection. A set of CLas-specific LAMP primers, loop forward (LF), loop backward (LB), forward outer primer (F3), backward outer primer (B3), forward internal primer (FIP), and backward internal primer (BIP) were designed based on the nucleotide sequence of the nrdB gene using Primer Explorer software (https://primerexplorer.jp/e/) (Table 1).

Nucleotide sequence of primers for rt-LAMP to detect CLas by targeting five copy nrdB gene
Optimization of rt-LAMP
The optimization of the rt-LAMP was conducted with Bst 2.0 WarmStart DNA polymerase (NEB, Ipsiwich, MA, USA) using the NEB LAMP protocol as a guideline. The 25 μl rt-LAMP reaction included 1 μM EvaGreen dye (Biotium, Fremont, CA, USA) to facilitate the real-time detection of the amplified LAMP targets on Bio-Rad CFX96 real-time PCR (Hercules, CA, USA) from which the relative fluorescence unit (RFU) was monitored at every 30 s or 1 min (i.e., one rt-LAMP cycle equals to 30 s or 1 min). Since the output data on a Bio-Rad CFX96 was generated as the number of qPCR “threshold cycle (Ct)”, we use the term “threshold time (Tt)” for the cycle number of rt-LAMP assay, which can be considered as an equivalent of qPCR “threshold cycle (Ct)”. In addition, as the RFU was monitored at every 30 s or 1 min depending on the experiment, the unit of Tt was indicated in each figure and table for clarification.
The optimum temperature for the rt-LAMP assay was selected after setting a gradient rt-LAMP assay with the temperature gradient between 50–68°C. Then, the optimum molar ratio of LAMP primers, FIP/BIP: F3/B3: LF/LB, was examined while keeping the molar concentration of each internal primer (FIP/BIP) at 1.6 μM. The molar ratios of primers examined were as follows: FIP/BIP:F3/B3:LF/LB = (1) 1.6 μM:0.4 μM:0.2 μM, (2) 1.6 μM:0.4 μM:0.4 μM, (3) 1.6 μM:0.4 μM:0.8 μM, and (4) 1.6 μM:0.2 μM:0.8 μM. The optimum MgSO4 concentration was screened by testing rt-LAMP assay with 2 mM, 4 mM, 6 mM, and 8 mM of MgSO4.
The optimized rt-LAMP reaction used in the remaining studies contained 6 mM MgSO4, 1.4 mM dNTPs, 1.6 μM of each internal primer, FIP and BIP, 0.4 μM of each outer primer, F3 and B3, 0.8 μM of each loop primer, LF and LB, 8U Bst2.0 WarmStart DNA polymerase and 1 μM Eva Green in 25 μl rt-LAMP reaction.
Quantitative real-time PCR
The qPCR for the detection of CLas was conducted using the primer-probe set, called RNR that targets nrdB gene (Zheng et al., 2016) in various tissue samples. The psyllid samples were tested by qPCR using a primer-probe set (HLBaspr) that targets CLas 16S rDNA (Li et al., 2006) together with a psyllid primer-probe set that targets glycoprotein (WG) gene as an internal control (Li et al., 2008).
qPCR was performed in 25 μl reactions containing 2 μl template DNA, 6 mM of MgCl2, 0.24 mM dNTPs, 0.24 μM of forward and reverse primer, 0.12 μM of probe, and 1 U Platinum Taq DNA Polymerase (Invitrogen, Carlsbad, CA, USA). The qPCR was conducted by 1 cycle of denaturation for 2 min at 95°C, followed by 40 cycles of 95°C for 10 s and 58°C for 40 s. The qPCR threshold cycle (Ct) value below 37 was considered a positive for CLas.
Estimation of CLas detection sensitivity of rt-LAMP compared to qPCR
For the estimation of CLas detection limit of rt-LAMP compared to qPCR, a standard curve was generated using a recombinant plasmid DNA containing a 204 bp-long partial fragment of CLas nrdB gene after linearizing the recombinant plasmid DNA with RsaI (NEB). The digested DNA was analyzed by agarose gel electrophoresis from which the linearized plasmid DNA was eluted using Zymoclean Gel DNA Recovery Kit (Zymo Research, Irvine, CA, USA). A series of tenfold dilution (up to 10−9 dilutions) of the linearized DNA was prepared from which a standard curve was generated by qPCR using an RNR primer set (Zheng et al., 2016).
Results
Optimization of rt-LAMP for CLas detection by targeting five copy nrdB gene
CLas-positive DNA samples confirmed by qPCR were used as a template for the optimization of the rt-LAMP assay.
Optimization of rt-LAMP reaction temperature and primer concentration
A gradient rt-LAMP reaction was conducted with a temperature gradient between 50°C and 68°C. The RFU of the rt-LAMP reaction was monitored every 1 min (i.e., Tt = 1 equals to 1 min). The Tt value of the rt-LAMP obtained at 67°C and 68°C were 9.39 and 9.21 respectively, which were the two lowest Tt values obtained from the gradient rt-LAMP assay (Fig. 1). For primer optimization, the reaction temperature of 67°C was selected. Among four different molar ratios of LAMP primers examined (Fig. 2), the rt-LAMP reaction with 1.6 μM of each internal primer, FIP and BIP, 0.4 μM of each external primer, F3 and B3, and 0.8 μM of each loop primer, LF and LB had the lowest Tt value of 7.22, and was selected as the primer concentration for the remainder of the studies (Fig. 2).

A gradient real-time loop-mediated isothermal amplification (rt-LAMP) reaction with a temperature gradient from 50°C to 68°C. ‘Candidatus Liberibacter asiaticus’ (CLas)-positive leaf DNA was used as a template for this assay. One rt-LAMP cycle equals to 1 min.

Optimization of the real-time loop-mediated isothermal amplification (rt-LAMP) primer concentration. The molar ratios of primers examined were FIP/BIP:F3/B3:LF/LB = (A) 1.6 μM:0.4 μM:0.8 μM; (B) 1.6 μM:0.4 μM:0.4 μM; (C) 1.6 μM:0.4 μM:0.2 μM; (D) 1.6 μM:0.2 μM:0.8 μM. One rt-LAMP cycle equals to 1 min.
Optimization of MgSO4 concentration
A series of rt-LAMP reactions with four different MgSO4 concentrations, 2 mM, 4 mM, 6 mM, and 8 mM MgSO4 were conducted, from which the rt-LAMP reaction conducted with 4 mM and 6 mM of MgSO4 had the lowest Tt values at 7.54 and 7.48, respectively (Fig. 3). To further examine the optimum MgSO4 concentration for the rt-LAMP assay, a standard curve was generated by qPCR for a quantification purpose using a linearized recombinant plasmid DNA containing 204 bp-long partial nrdB gene (Fig. 4). Then, the rt-LAMP reactions were repeated with 4 mM or 6 mM MgSO4 using another set of dilutions of the linearized recombinant plasmids. Table 2 showed that the rt-LAMP reactions with 6 mM MgSO4 had slightly lower Tt values than those with 4 mM MgSO4, from which 6 mM MgSO4 was selected as an optimum concentration for rt-LAMP reaction.
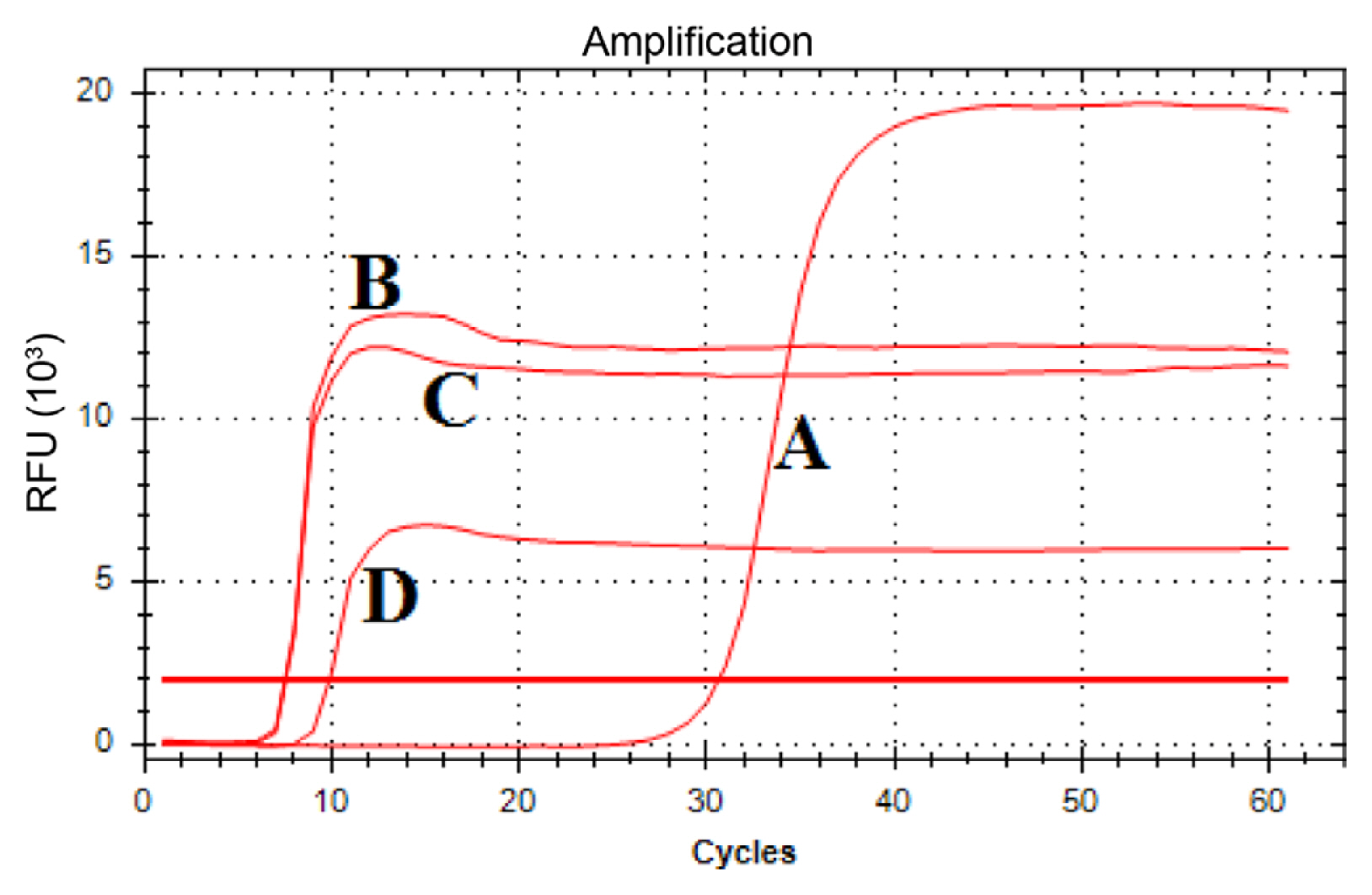
Optimization of the MgSO4 concentration for the real-time loop-mediated isothermal amplification (rt-LAMP) assay: 2 mM (A), 4 mM (B), 6 mM (C), and 8 mM (D) of MgSO4. One rt-LAMP cycle equals to 1 min.

Standard curve generated with a series of dilutions of a recombinant plasmid that has a partial fragment of ‘Candidatus Liberibacter asiaticus’ (CLas) nrdB gene. Standard curve and linear regression were indicated on the plot (A). The dilution factor, the Log10 copy number of targets in each dilution, and the quantitative real-time polymerase chain reaction Ct value were shown in the table (B). aThe Ct standard deviation was obtained from triplicates.

Comparison of the rt-LAMP data obtained with 4 mM and 6 mM MgSO4
Based on the optimization data described above, the remaining studies were conducted with 6 mM MgSO4, 1.4 mM dNTPs, 1.6 μM of FIP/BIP, 0.4 μM of F3/B3, 0.8 μM of LF/LB, 8 U Bst2.0 WarmStart DNA polymerase, 2 μl DNA, and 1 μM Eva Green in 25 μl rt-LAMP reaction.
Estimation of CLas detection limit of rt-LAMP
Table 2 revealed that the rt-LAMP reactions with low target molecules (i.e., <~2.5 Log10 copies) showed the delayed detection (i.e., increased Tt). To estimate the detection limit of the optimized rt-LAMP reaction compared to qPCR, the rt-LAMP reaction was re-conducted with the triplicates of the diluted linearized plasmid. Table 3 showed that although the rt-LAMP detected the target molecule as low as 2.05 Log10 copies (~1.1 × 102 copies) in the reaction, there was substantially increased variation between replicates (standard deviation >10) in the detection time (Tt) of the samples with less than 2.5 Log10 target molecules. Since this detection limit was obtained with a recombinant plasmid (Table 3), the experiment was repeated with CLas-positive root DNA fraction as described below in order to investigate the detection limit of the rt-LAMP with CLas field samples.

Comparison of CLas detection limit of the rt-LAMP assay with that of qPCR using series of diluted linearized plasmid DNA
CLas-positive root DNA fraction was serially diluted 1:5 with healthy root DNA fractions, which was used as a template for the rt-LAMP reaction. Table 4 showed that while qPCR detected ~1.05 of Log10 target copies present in the sample with Ct value of 36.66, only the DNA samples containing ≥~2.6 of Log10 target copies tested positive by the rt-LAMP assay with a minor variation in the detection time between replicates, indicating that the detection limit of the rt-LAMP assay is 2.6 Log10 target copies (~3.98 × 102 copies).

Comparison of the detection limit of rt-LAMP assay with qPCR using a serially diluted CLas-positive root DNA with heathy root DNA fraction
The efficacy of the rt-LAMP assay for CLas detection using various tissue samples
To evaluate the effectiveness of the rt-LAMP assay for the detection of CLas in field samples, various tissue samples were collected from trees planted in a commercial orchard located in Edinburg, Texas. These samples included 67 leaf, 33 bark, and 58 fibrous root samples. In addition, 45 psyllids were also collected from a field in Weslaco, TX, USA.
Among 67 leaf samples, 58 samples tested positive for CLas by qPCR, and 56 by rt-LAMP assay showing 96.6% detection rate compared to qPCR (Table 5). Similarly, among 33 bark DNA extracts, 32 samples tested positive for CLas by qPCR, and 31 by the rt-LAMP assay showing a 96.8% detection rate compared to qPCR. In the case of fibrous root samples, while 46 of 58 samples tested positive for CLas by qPCR, only 26 samples were tested positive by the LAMP assay showing a 56.5% detection rate compared to qPCR (Table 5). Of 46 CLas-positive root samples confirmed by qPCR, 20 root samples that were tested negative by the rt-LAMP assay had Ct value greater than 31, indicating that the estimated Log10 target copy number in these samples was less than ~2.6 that was the detection limit of the rt-LAMP assay (Table 4). When only those root samples with Ct value below 30 were selected for comparison, the detection rate of CLas-positive root samples by the rt-LAMP assay increased to 96.1% (data not shown). The rt-LAMP reaction conducted with psyllid samples showed 97.7% detection rate compared to qPCR (Table 5).

CLas detection rate of rt-LAMP compared to qPCR using various tissue samples collected from field
Field applicability of the rt-LAMP assay for CLas detection
In order to examine the field applicability of the rt-LAMP assay, the purified DNA fractions prepared from 18 leaf, bark, and fibrous root samples, collected from grapefruit trees grafted on sour orange rootstock, were tested with rt-LAMP on a portable device (BioRanger, Diagenetix Inc., Honolulu, HI, USA). Then, the data was compared to the results obtained with qPCR as well as with the rt-LAMP conducted on a real time PCR system (Table 6). Table 6 showed that the CLas detection on a portable device by rt-LAMP was achieved in less than 15 min (range, 8.5–12.5 min). The CLas detection rate from leaf and bark samples by rt-LAMP assays conducted on the real-time and the portable devices was comparable to that of qPCR. On the other hand, the rt-LAMP detected CLas from 11 out of 13 root samples that were tested positive by qPCR (Table 6). Those two root samples tested negative by rt-LAMP had Ct values of 33.56 and 33.21 of which estimated Log10 target copy numbers were 1.9 and 2.0, respectively, that were below the detection limit of the rt-LAMP assay (Table 6). Based on the rt-LAMP data obtained with the root sample with Ct value of 31.31, the rt-LAMP can detect CLas from the field sample with as low as ~2.5 Log10 copies of target molecule (Table 6). In addition, Table 6 showed that the rt-LAMP data obtained with a portable device was consistent with the data obtained with the rt-LAMP conducted on the real-time PCR system, which confirmed the field applicability of the rt-LAMP assay when it is coupled with a portable device.

Comparison of CLas detection by qPCR and the rt-LAMP assays on a real-time PCR and a portable device
Discussion
As one of the most devastating diseases of citrus, causing significant economic damage, monitoring HLB incidence is critical to disease management strategies. Under this circumstance, the most pursued measure towards determining HLB incidence has been visual surveys in the field for trees with HLB symptoms. The infected trees develop visible symptoms on leaves at the later stage of infection, varying from six months to one or more years after natural infection, which makes early HLB diagnosis difficult to achieve (da Graça et al., 2016; Gottwald et al., 2007). At present, HLB diagnosis is performed by the following multiple steps: (1) field survey by scouts for HLB symptoms, (2) sending the leaves with the HLB-like symptoms to the lab equipped with proper lab instruments, (3) DNA extraction and qPCR for HLB diagnosis and (4) reporting the test result to the grower. Although the qPCR-based HLB diagnosis is the most sensitive and reliable method for HLB diagnosis, it is time-consuming. As an effort to diagnose the disease at the early stage, HLB diagnosis using fibrous root samples has been reported as an efficient method for earlier disease diagnosis than with leaf tissue (Braswell et al., 2020; Park et al., 2018). Braswell et al. (2020) showed that HLB spread in young plantings took place extremely fast both in Florida and Texas, where the percentage of CLas-positive trees in the field increased in a year from ~22% to ~90% in Florida and ~18% to ~70% in Texas. Such rapid spread emphasizes the need for a field tool for a quick CLas detection, especially for the trees with HLB-like symptoms on the canopy.
The current study adopted a novel nucleic acid amplification method, LAMP, for the detection of CLas, a causal agent of HLB disease in citrus. As the operation of isothermal assays takes place at a constant temperature, the CLas detection by LAMP assay is relatively simple compared to qPCR-based methods that require thermocycling for target DNA amplification. Although LAMP assays for CLas detection have been previously reported by targeting different CLas genomic loci (Choi et al., 2018; Keremane et al., 2015; Okuda et al., 2005; Rigano et al., 2014), the acceptance of these assays for CLas detection in the field has been very limited. In addition, the CLas detection limit of these LAMP assays varied depending on the target region of CLas genome (Choi et al., 2018; Keremane et al., 2015; Okuda et al., 2005; Rigano et al., 2014). One potential explanation for the varied detection sensitivity between different LAMP assays could be due to the difference in the copy number of the target region in the CLas genome. Here, we developed a rt-LAMP assay by targeting CLas nrdB gene, a gene with five copies in the CLas genome (Zheng et al., 2016), which could provide an improved detection sensitivity due to its higher target copy number.
The rt-LAMP method, described here, can detect CLas in various tissue samples (i.e., leaf, bark, root, and psyllid) with target molecules as low as ~2.6 Log10 copies. Although the CLas detection limit of the rt-LAMP is lower than that of qPCR, the data obtained with leaf, bark and psyllids collected from the field showed that rt-LAMP has >96% CLas detection rate when it was compared to qPCR data. In addition, a preliminary data obtained with asymptomatic leaves confirmed that the rt-LAMP assay does not cross-react with non-target microbes residing in the citrus leaf samples (Supplementary Fig. 1). We also confirmed that the rt-LAMP assay can be conducted with plant crude extracts (Supplementary Fig. 2). These data indicated that the rt-LAMP assay developed in the study can be deployed in the field for quick CLas detection using plant crude extracts on a portable device.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Electronic Supplementary Material
Supplementary materials are available at The Plant Pathology Journal website (http://www.ppjonline.org/).