Genetic Variation of Monilinia fructicola Population in Korea
Article information

Abstract
Brown rot disease, caused by Monilinia spp., poses a significant threat to pome and stone fruit crops globally, resulting in substantial economic losses during pre- and post-harvest stages. Monilinia fructigena, M. laxa, and M. fructicola are identified as the key agents responsible for brown rot disease. In this study, we employed the amplified fragment length polymorphism (AFLP) method to assess the genetic diversity of 86 strains of Monilinia spp. isolated from major stone fruit cultivation regions in South Korea. Specifically, strains were collected from Chungcheong, Gangwon, Gyeonggi, Gyeongsang, and Jeolla provinces (-do). A comparative analysis of strain characteristics, such as isolation locations, host plants, and responses to chemical fungicides, was conducted. AFLP phylogenetic classification using 20 primer pairs revealed the presence of three distinct groups, with strains from Jeolla province consistently forming a separate group at a high frequency. Furthermore, M. fructicola was divided into three groups by the AFLP pattern. Principal coordinate analysis and PERMANOVA were applied to compare strain information, such as origin, host, and fungicide sensitivity, revealing significant partition patterns for AFLP according to geographic origin and host plants. This study represents the utilization of AFLP methodology to investigate the genetic variability among M. fructicola isolates, highlighting the importance of continuous monitoring and management of variations in the brown rot pathogen.
Worldwide, major fungal pathogens associated with stone fruit diseases include Alternaria spp., Botrytis cinerea, Colletotrichum spp., Monilinia spp., Phomopsis vexans, Phyllosticta circumscissa, Podosphaera tridactyla, and Septoria cerasina (Martini and Mari, 2014; Oh et al., 2017). Among them, Monilinia is a genus of fungi in the Sclerotiniaceae family, known to cause significant losses in various Rosaceae fruits and stone fruits (Oh et al., 2017). Brown rot disease caused by fungi in the Monilia genus inflicts significant economic damage during pre-harvest, post-harvest, storage, and transportation (Rural Development Administration, 2007). Brown rot disease manifests initially as small brown spots on the fruit surface. As the disease progresses, these spots enlarge into sunken lesions, leading to a reduction in fruit yield and compromising its marketability (Choi et al., 2016; Hu et al., 2011).
The genus Monilinia is responsible for the brown rot disease. Globally, Monilinia fructicola, M. fructigena, and M. laxa are the most common species all over the world (De Miccolis Angelini et al., 2018; Shim et al., 2007). Among them, M. fructicola is predominantly reported in the United States, Australia, New Zealand, and some parts of Europe and Japan. M. fructigena is prevalent in Europe and Japan, while M. laxa is documented to be distributed in the United States, Australia, Europe, and Japan (Fulton and Brown, 1997; Hu et al., 2011; Shim et al., 2007). In particular, in Korea, M. fructicola has been reported to cause brown rot disease in fruits such as peaches, plums, apricots, and plum cherries (Zhu et al., 2005). The control of stone fruit brown rot disease depends on chemical and cultivation methods (Martini and Mari, 2014). In terms of cultural control, practices involve removing dead leaves for improved ventilation, and early removal and incineration of infected branches and fruits (Lee and Kwak, 2023). Another approach is plowing the soil in the outbreak area until early spring to bury the pathogen deep within the soil. Chemical control involves the application of various fungicides during the onset of the disease (Hrustić et al., 2018).
To control brown rot disease, synthetic fungicides, including methyl benzimidazole carbamates (MBC), sterol biosynthesis inhibitors, succinate dehydrogenase inhibitors (SDHI), demethylation inhibitors (DMI), and quinone outside inhibitors (QoI), constitute the primary means of pathogen control. MBC-class fungicides such as benomyl and thiophanate-methyl are commonly used. Additionally, there are DMI group fungicides like tebuconazole and bitertanol, QoI group fungicides like pyraclostrobin, and SDHI group fungicides such as boscalid (Jones and Ehret, 1976; Lesniak et al., 2021; Usman et al., 2021). However, repeated use of fungicides has diminished the sensitivity of pathogens to the fungicides, reducing the effectiveness of the fungicides. Consequently, this has led to the emergence of resistant strains against the fungicides (Kim, 2000). In 1978, the first instance of resistance in brown rot pathogens to dicarboximide and benzimidazole fungicides was reported (Sztejnberg and Jones, 1978). For over 30 years, DMI fungicides have been used for the control of brown rot. During this period, variants of brown rot exhibiting resistance to DMI fungicides, along with the associated genetic factor ‘Mona’, a 65-bp sequence element that regulated MfCYP 51 expression, have been reported (Luo and Schnabel, 2008; Schnabel et al., 2004). In Korea, resistance to fungicides for brown rot has been reported since the 1970s. Currently, resistance to benzimidazole-class fungicides such as carbendazim, benomyl, and thiophanate-methyl, as well as dicarboximide-class fungicides like iprodione and procymidone, has been documented (Lim et al., 1998).
Disease symptoms did not allow for the differentiation of Monilinia species infecting both stone and pome fruits. Therefore, achieving a diagnosis at the species level often necessitates the evaluation of a combination of traits, encompassing colony morphology in vitro and DNA sequence characteristics (Côté et al., 2004; Gell et al., 2007; Lane, 2002). Analyzing the genetic structure of Monilinia populations yields insights into the origins of genetic variation, isolated regions, fungicide resistance groups, and host plant associations (Fulton et al., 1999). The internal transcribed spacer region (ITS) of rDNA exhibits sufficient resolution (5–13 nucleotide substitutions) to distinguish these three Monilinia species, but it provides little information on intraspecies variation (van Leeuwen et al., 2002). Investigations into phylogenetic relationships and genetic variations based on morphological and cytological traits can be influenced by environmental factors (Basu et al., 2004; Kumar et al., 2009; Sammour et al., 2013). Therefore, molecular markers have emerged as powerful tools for assessing genetic diversity and phylogenetic relationships in fungi.
Therefore, to achieve successful and efficient disease management within the same species groups, it is necessary to monitor genetic diversity. Amplified fragment length polymorphism (AFLP) is a suitable method to evaluate population variation and diversity at the genomic DNA levels (Vos et al., 1995). Among various molecular markers, the AFLP molecular marker system has been reported to successfully reveal genetic variations among or within populations that cannot be distinguished through morphological or other molecular tools (Majer et al., 1996; Purwantara et al., 2000; Schnieder et al., 2001). This study represents the first application of the AFLP technique to investigate the causal agent of brown rot disease, M. fructicola, in Korea. This study was designed to observe the genetic diversity of M. fructicola in Korea. AFLP analysis with principal coordinate analysis (PCoA) was conducted on 86 strains, utilizing isolated regions, chemical fungicide responsiveness results, and host plants for sampling to evaluate the relationship between the genetic diversity and characteristics of each strain.
Materials and Methods
Pathogen isolates and culture conditions
A total of 86 single-spore isolates of M. fructicola were included in the study. The isolates were collected from a wide range of hosts comprising pome and stone fruits from various locations in Korea (Table 1, Fig. 1): 58 isolates from peach (Prunus persica), 10 from apricot (Prunus armeniaca), 3 from Japanese plum (Prunus salicina), 3 from plum (Prunus mume), 9 from pluot (Prunus salicina × armeniaca), and 3 from cherry (Prunus pauciflora Bunge). All collected isolates were cultured from sporodochia originating from various parts of infected host tissues. Isolates were maintained on potato dextrose agar (PDA; potato dextrose 20 g, agar 15 g per liter) medium in Petri dishes in the dark condition at 25°C. For long-term storage, the isolated fungi were preserved at −80°C with a solution containing 20% glycerol and 17% skim milk.
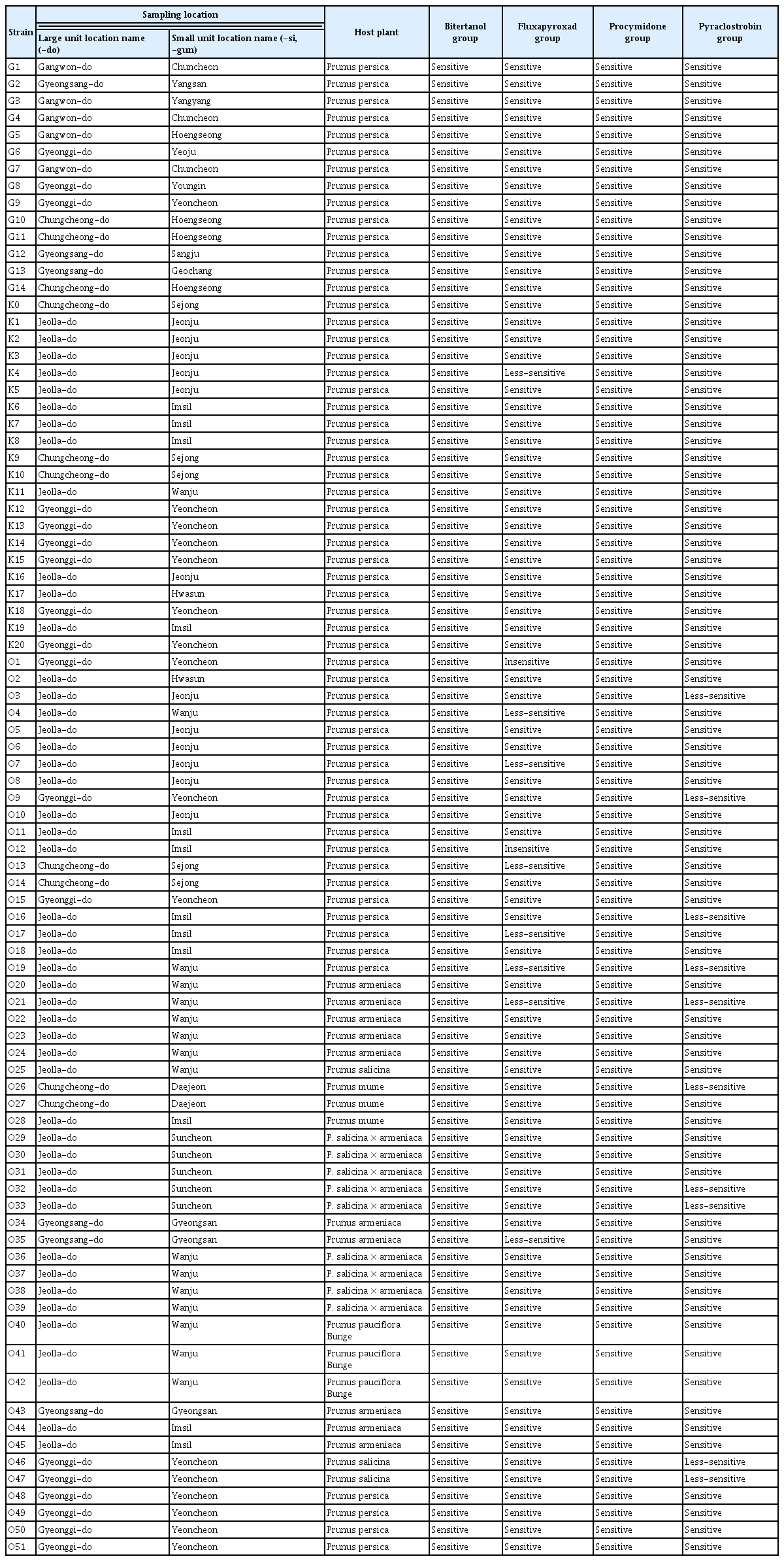
Monilinia fructicola isolates information

Site information where strains are isolated. Small district unit (-si, and -gun) names of the site are described by points and large unit (-do) names are described by the colors of points. In all sites, there are a total of 16 distinct regions.
Genomic DNA extraction and identification of the pathogen
For molecular identification of the isolated pathogens, DNA extraction was carried out. The isolated pathogens were inoculated onto one-fifth strength PDA (potato dextrose 4.8 g, agar 15 g per liter) and cultivated at 25°C for 10 days. The cultivated pathogens were subjected to DNA extraction using the cetyltrimethylammonium bromide (CTAB) method. Fungal spores and mycelium were transferred to a 1.5-ml E-tube containing 500 μl of CTAB (CTAB 2.0% (w/v), 100 mM Tris-HCl, pH 8.0, 20 mM EDTA pH 8.0, 1.4 M NaCl), and 8 μl of proteinase K solution (20 mg/ml) was added. The mixture was then crushed, and a 30 min reaction was carried out 65°C. After the reaction, it was cooled at room temperature for 10 min, followed by the addition of PCI (phenol:chloroform:isoamyl alcohol, 24:25:1) 500 μl, mixed by inverting 5 to 7 times. The mixture was centrifuged at 12,470 ×g for 10 min to remove proteins. The supernatant was transferred to a new E-tube, and 400 μl of isopropanol was added. After centrifugation at 12,470 ×g for 5 min at 4°C, the supernatant was removed. Finally, 70% ethanol (500 μl) was added, centrifuged for 5 min, and the supernatant was removed. The pellet was air-dried for 1–2 h. The DNA concentration was measured by adding 20 μl of TE buffer to the E-tube and using the NanoDrop 2000C spectrophotometer (Thermo Scientific, Waltham, MA, USA). The extracted DNA was used for the amplification of the ITS, TEF1-α, β-tubulin, and cytB regions. The primers used were ITS1 (5′-TCC GTA GGT GAA CCT GCG G-3′)/ITS4 (5′-TCC TCC GCT TAT TGA TAT GC-3′) for the ITS region, EF1-728F (5′-CAT CGA GAA GTT CGA GAA GG-3′)/EF1-986R (5′-TAC TTG AAG GAA CCC TTA CC-3′) for TEF1-α, TubF1 (5′-ATG CGT GAG ATT GTA CGT AT-3′)/TubR1 (5′-GTACCAATGCAAGAAAGCCT-3′) for β-tubulin, and colaexon3-fwd (5′-TTT ACC TTA CGG TCA AAT GAG CCT-3′)/colaexon4-rev (5′-AAC TCA ACA ATA TCA CCT CCA ATT CAT-3′) for cytB (Fischer et al., 2017; Hily et al., 2011; Poniatowska et al., 2021; White et al., 1990). The sequence analysis was conducted by Cosmogentech (Seoul, Korea), and the obtained sequences were identified using the NCBI nucleotide blast search program.
Amplified fragment length polymorphism (AFLP)
EcoRI (10 unit) and MspI (10 unit) were used to digest 200 ng of genomic DNA in a 20 μl reaction at 37°C for 5 h. The digested DNA was ligated to EcoRI adapter (0.1 μM: 5′-GACGATGAGTCCTGAT-3′, 5′-CGATCAGGACTCATC-3′) and MspI adapter (0.1 μM: 5′-GACGATGAGTCCTGAG-3′, 5′-CGCTCAGGACTCAT-3′) using 40 units of T4 DNA ligase, and 0.2 mM ATP in a 50 μl reaction at 37°C for 3 h. Following a 20-fold dilution of the ligated product, nonselective amplification was carried out using EcoRI+0 (5′-GACTGCGTACCAATTC-3′) and MspI+0 (5′-GATGAGTCCTGAGCGG-3′) primers. The thermal conditions included pre-denaturation at 94°C for 2 min, 20 amplification cycles (94°C for 30 s, 56°C for 1 min, and 72°C for 1 min), and final extension at 72°C for 90 s. The PCR products from the pre-amplification step were diluted 10-fold, and 1 μl of the dilution was used for selective amplification with the same core primers as the first amplification, along with two arbitrary (selective) nucleotides at the 3′ end. Selective reactions were performed in a 20 μl PCR reaction mix with the same components as the pre-amplification step, using 0.3 U of Taq polymerase and 50 ng of each selected primer. EcoRI primers were FAM labeled at their 5′ end for automated laser fluorescence analysis. The reaction involved an initial touch-down protocol: 12 cycles of primer-sensitive amplification (94°C for 30 s, 65°C decreased by 0.7°C during each cycle for 30 s, and 72°C for 60 s), 23 normal cycles (94°C for 30 s, 56°C for 30 s, and 72°C for 60 s), and a final elongation at 72°C for 10 min. The FAM-labeled amplicon was analyzed using a DNA Analyzer ABI3730XL (Perkin Elmer, Waltham, MA, USA) at the National Instrumentation Center for Environmental Management. From an initial screening of 40 different primer combinations on the fungal isolates, 20 primer combinations (comprising 4 EcoRI [E] and 10 MspI [M] primers) were selected based on amplification profile intensity, number of bands, and indicative polymorphism. M-TA, M-GT, M-TC, M-CT, M-TG, and M-AG were combined with E-GA; E-GA was combined with M-CG, M-CT, M-AT, M-TA, M-AG, M-TC, and M-GT; E-GT was combined with M-CG, M-GA, and M-GC; and E-TC was combined with M-TA, M-GA, M-AG, and M-CG (Gril et al., 2010).
Statistical analysis
The FAM-labeled DNA fragments generated from the DNA analyzer were obtained as fragment analysis (FSA) files. Fragment information within the FSA files was managed using the Fragman package (version 1.0.9) and analyzed with stats (version 4.3.2), ape (analysis of phylogenetic and evolution, version 5.7.1), nortest (version 1.0.4), vegan (version 2.6.4), and pairwiseAdonis (version 0.4.1) package in R (version 4.3.2). AFLP was evaluated based on sensitivity, geographical separation, and host plant for previously studied fungicides including bitertanol, pyraclostrobin, procymidone, and fluxapyroxad. The reported raw measurements involved the calculation of half-maximal effective concentration (EC50) for fungicides using a three or four-parameter log-logistic model in the drc package (version 3.0.1) in R. The log-logistic model and EC50 results were included in the supplementary data (data not shown). The results were visualized using ggplot2 (version 3.4.2) and ggpubr (version 0.6.0) packages in R.
Results and Discussion
Phylogenetic analysis of M. fructicola isolates nationwide
To obtain information based on pathogen variation, this study was designed to investigate genetic diversity using 86 of the pathogens isolated from stone fruit cultivation in Korea (Fig. 1) and examine the relationship between AFLP and strain characteristics. As a result, a total of 1,482 well-defined AFLP bands were amplified. Primer combination E-GA+ M-CG, M-CT, M-AT, M-TA, M-AG, M-TC, and M-GT amplified the highest number of fragments (379). On the other hand, the primer combination E-GT+ M-CG, M-GA, and M-GC showed the fewest bands (28). On average, 304.92 AFLP markers were amplified per primer combination in the size range of 40–500 bp. The fragment patterns were utilized to construct a phylogenetic tree using Bray-Curtis distance and the Weighted Pair Group Method with Averaging (WPGMA) hierarchical clustering. All four primer combinations were grouped into three major clusters based on a phylogenetic distance of 0.3, and isolates from Jeolla province show a high frequency in all clusters (Figs. 2 and 3). In particular, distinct patterns were observed between strains isolated from Jeolla province and those from other regions, especially with the primer combinations E-GA+M-CG, M-CT, M-AT, M-TA, M-AG, M-TC, M-GT (Fig. 2), and E-TC+M-TA, M-GA, M-AG, G-CG (Fig. 3). Furthermore, when comparing AFLP banding patterns obtained from an AFLP study on M. laxa, a causative agent of brown rot disease, previously reported by Gril et al. (2008), completely different electrophoretic profiles were observed. This suggests that the AFLP technique is a suitable tool for studying closely related groups of brown rot pathogens.
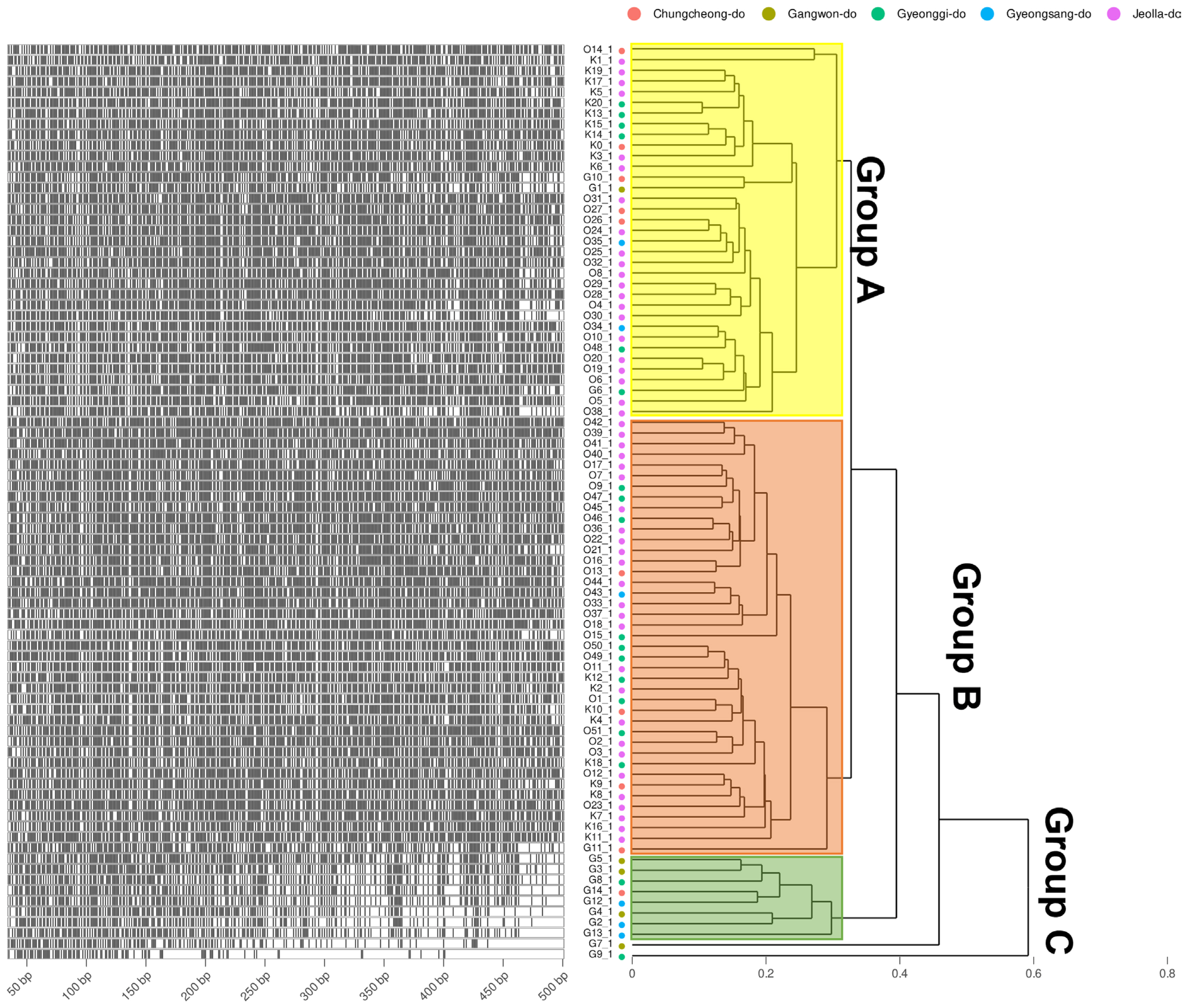
DNA fragments of amplified fragment length polymorphism and the fragment phylogeny of E-GA + M-CG, CT, AT, TA, AG, TC, GT primer combination. The Left of the plots shows the specifically amplified genomic DNA length information after the DNA fragmentation. Phylogeny on the right is calculated by the Bray-Curtis distance of the length and Weighted Pair Group Method with Averaging (WPGMA) clustering. Four different amplification profiles are shown when E-GA + M-CG, CT, AT, TA, AG, TC, and GT primer combinations were used. The group displayed by yellow, orange, and green translucent rectangles is determined based on 0.3.
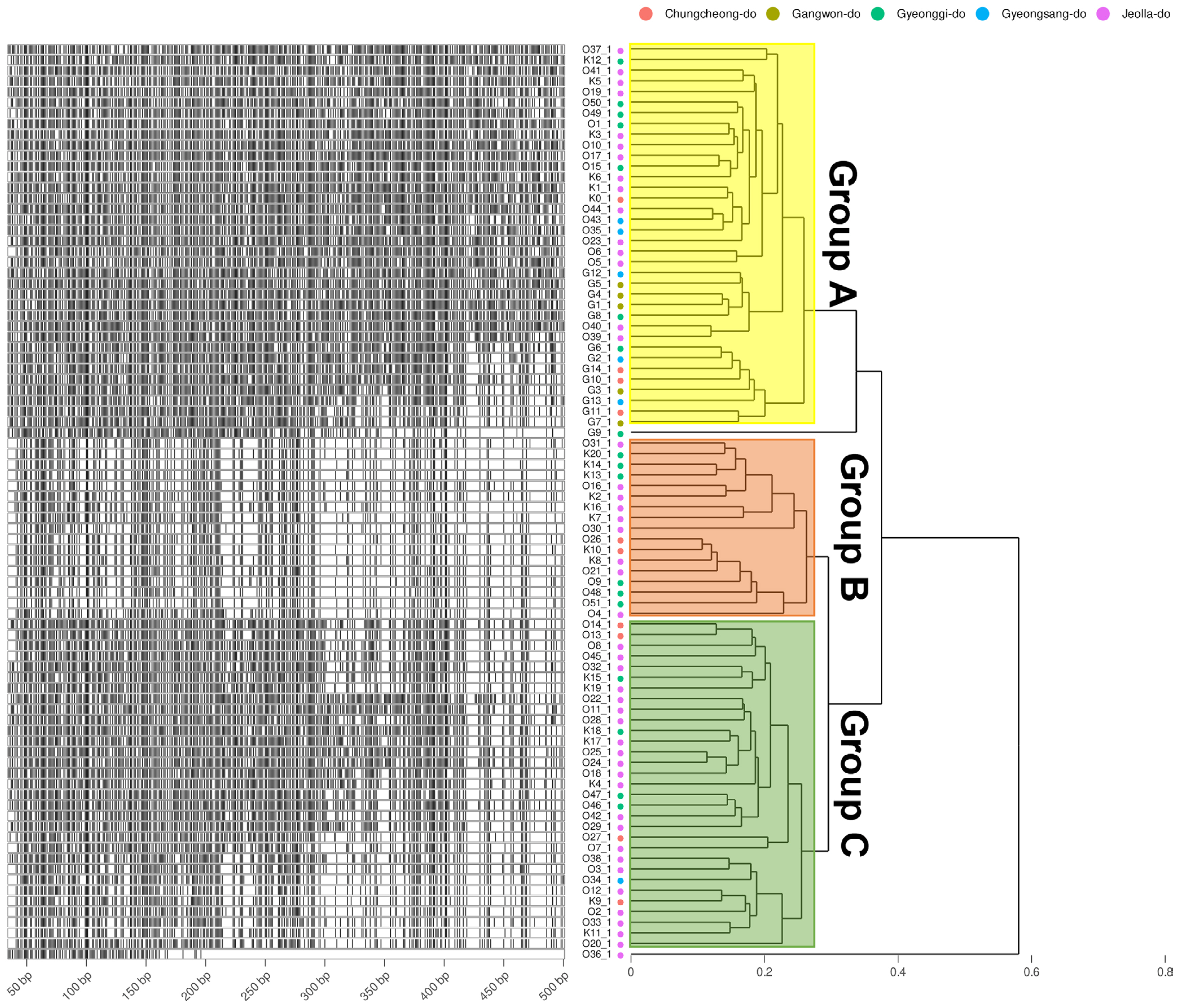
DNA fragments of amplified fragment length polymorphism and the fragment phylogeny of E-TC + M-TA, GA, AG, CG primer combination. The Left of the plots shows the specifically amplified genomic DNA length information after the DNA fragmentation. Phylogeny on the right is calculated by the Bray-Curtis distance of the length and Weighted Pair Group Method with Averaging (WPGMA) clustering. Four different amplification profiles are shown when E-TC + M-TA, GA, AG, and CG primer combinations were used. The group displayed by yellow, orange, and green translucent rectangles is determined based on 0.3.
PCoA of M. fructicola populations
In order to analyze the relationship more thoroughly between AFLP analysis and characteristics, we conducted a dimensional analysis using PCoA on primer combinations E-GA+M-CG, M-CT, M-AT, M-TA, M-AG, M-TC, M-GT, and E-TC+M-TA, M-GA, M-AG, G-CG pattern combinations (Figs. 4 and 5). Then PERANOVA observed whether the distribution of the pattern of the fragment varies by characteristic (Tables 2 and 3, Figs. 4 and 5). For the category of fungicides EC50 were based on their quartiles (data not shown). Subsequently, PERMANOVA and pairwise PERMANOVA results (Tables 2 and 3), calculated based on point distribution, indicated significant differences in distribution (PERMANOVA, P < 0.10; pairwise PERMANOVA, Padj < 0.10) at the large-scale locational unit (province). The results from Tables 2 and 3 diverge from the findings of the AFLP pattern analysis. While the AFLP pattern analysis indicates a difference between Jeolla province and other regions, the results from Tables 2 and 3 suggest a significant difference between Gangwon province and other regions. Moreover, significant distribution differences were identified among groups within the AFLP phylogeny (PERMANOVA, P < 0.001; pairwise PERMANOVA, Padj < 0.001). However, no significant differences were observed between host plants and fungicide resistance groups (Figs. 4 and 5). In previous studies, isolates from diverse geographical locations (Spain, New Zealand, and Japan) from the same hosts were clustered, also suggesting a potential correlation with host specificity (Gril et al., 2010). Additionally, in another study, strains isolated from Virginia and Washington in the United States, though geographically separated, were considered to share a common origin based on being collected from the same host plant (Fulton et al., 1999). This suggests that, despite the diversity of host plants in Korea, significant differences occurred depending on the geographical separation.

The genomic DNA fragment principal coordinate analysis results of E-GA + M-CG, CT, AT, TA, AG, TC, GT primer combination. Point meaning each Monilinia fructiocola strain is flat. The graph is organized based on amplified fragment length polymorphism (AFLP) phylogeny group, small unit location (-si, -gun), large unit location (-do), host plant, fluxapyroxad sensitive group, pyraclostrobin sensitive group, and the ellipses represent the extent to which 95% of points are distributed. (A) AFLP phylogeny group. (B) Small unit location group. (C) Large unit location group. (D) Host plant groups. (E) Fluxapyroxad sensitive group. (F) Pyraclostrobin sensitive group. Phylogeny groups (except for no group), PERMANOVA, P = 1e-05, Location (-do), PERMANOVA, P = 7e-05.

The genomic DNA fragment principal coordinate analysis results of E-TC + M-TA, GA, AG, CG primer combination. Point meaning each Monilinia fructiocola strain is flat. The graph is organized based on amplified fragment length polymorphism (AFLP) phylogeny group, small unit location (-si, -gun), large unit location (-do), host plant, fluxapyroxad sensitive group, pyraclostrobin sensitive group, and the ellipses represent the extent to which 95% of points are distributed. (A) AFLP phylogeny group. (B) Small unit location group. (C) Large unit location group. (D) Host plant groups. (E) Fluxapyroxad sensitive group. (F) Pyraclostrobin sensitive group. Phylogeny groups (except for no group), PERMANOVA, P = 1e-05, Location (-do), PERMANOVA, P = 7e-05.

PERMANOVA and paired-wised PERMANOVA with FDR method of E-GA + M-CG, CT, AT, TA, AG, TC, GT primer combination

PERMANOVA and paired-wised PERMANOVA with FDR method of E-TC + M-TA, GA, AG, CG primer combination
This study investigated the genetic differentiation of M. fructicola, examining its influence based on cultivation regions, host plants, fungicide usage, and various environmental conditions. Different patterns were observed depending on the region, and significant differences were identified. However, the number of fungicide-resistant strains was limited. This highlights the need for further research, diverse approaches, and genetic diversity monitoring for sustainable agriculture.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Acknowledgments
This research was supported by the Rural Development Administration of Korea (RS-2022-RD010350).