Deep Sequencing Analysis of Apple Infecting Viruses in Korea
Article information
Abstract
Deep sequencing has generated 52 contigs derived from five viruses; Apple chlorotic leaf spot virus (ACLSV), Apple stem grooving virus (ASGV), Apple stem pitting virus (ASPV), Apple green crinkle associated virus (AGCaV), and Apricot latent virus (ApLV) were identified from eight apple samples showing small leaves and/or growth retardation. Nucleotide (nt) sequence identity of the assembled contigs was from 68% to 99% compared to the reference sequences of the five respective viral genomes. Sequences of ASPV and ASGV were the most abundantly represented by the 52 contigs assembled. The presence of the five viruses in the samples was confirmed by RT-PCR using specific primers based on the sequences of each assembled contig. All five viruses were detected in three of the samples, whereas all samples had mixed infections with at least two viruses. The most frequently detected virus was ASPV, followed by ASGV, ApLV, ACLSV, and AGCaV which were withal found in mixed infections in the tested samples. AGCaV was identified in assembled contigs ID 1012480 and 93549, which showed 82% and 78% nt sequence identity with ORF1 of AGCaV isolate Aurora-1. ApLV was identified in three assembled contigs, ID 65587, 1802365, and 116777, which showed 77%, 78%, and 76% nt sequence identity respectively with ORF1 of ApLV isolate LA2. Deep sequencing assay was shown to be a valuable and powerful tool for detection and identification of known and unknown virome in infected apple trees, here identifying ApLV and AGCaV in commercial orchards in Korea for the first time.
Introduction
Apple is an economically important fruit crop, covering an area of 19,313 ha with an annual production of about 583,000 tonnes in Korea (http://kostat.go.kr, 2015). Apple trees are affected by at least 12 viruses and virus like diseases, which cause significant economic losses (Németh, 1986; Nisar, 2013; Saade et al., 2000). Among these viruses, Apple chlorotic leaf spot virus (ACLSV), Apple stem grooving virus (ASGV), Apple stem pitting virus (ASPV), and Apple mosaic virus (ApMV) commonly occur in commercial apple orchards around the world. Previous surveys indicated that 47.6% of apple trees in Korea are infected by ACLSV, ASGV, and ASPV (Cho, 2015), but ApMV has not been detected in recent years in Korea. For the certification of apple plant material, rootstocks and cultivars have been tested along with other pathogens including four viruses, ACLSV, ASGV, ASPV, and ApMV; the use of certified healthy plant materials can prevent virus spread in commercial apple orchards.
Virus control is based mainly on prevention such as planting healthy propagation materials and the eradication of infected plants (Mathews, 2010; Rowhani et al., 1995). Therefore, reliable and sensitive virus detection methods are critical in successful screening for healthy plant materials (Sastry, 2013). Traditional virus detection assays such as woody indicators, ELISA, molecular hybridization, and RT-PCR are optimized for the detection of known viruses. However, novel and highly divergent viruses are not easily detected by the assays that depend on prior availability of specific antibodies or knowledge of sequences (Yozwiak et al., 2012). More recently, deep sequencing (or next generation sequencing) assay has provided a powerful alternative for the detection and identification of the total pathogen load in infected plants (the virome being the the totality of viral pathogens in an infected plant) without a priori knowledge (Li et al., 2012). Deep sequencing assay has been applied for virome diagnostics in fruit crops (Barba et al., 2014; Coetzee et al., 2010). In apples, Yoshikawa et al. (2012) identified ASGV, ASPV, ACLSV, Apricot latent virus (ApLV), Apricot pseudo-chlorotic leaf spot virus (ApPCLSV), and Peach chlorotic mottle virus (PCMV) from green crinkle disease of apple trees. In grapevines, Al Rwahnih et al. (2009, 2012) identified a novel Marafivirus, Grapevine Syrah virus-1 (GSyV-1) associated with grapevine syrah decline (2009), a novel circular DNA virus, Grapevine red blotch-associated virus (GRBaV) associated with grapevine red blotch disease and a novel Vitivirus, Grapevine virus F (GVF) from infected grapevine (2012). Giampetruzzi et al. (2012) discovered a novel RNA virus, Grapevine Pinot gris virus (GPGV) from infected grapevine. In Prunus spp., Candresse et al. (2013) identified Plum pox virus, Prunus necrotic ringspot virus and novel viral agents from prunus materials and a Little cherry virus 1 (LChV1) isolate associated with Shirofugen stunt disease syndrome of cherry plants. These previous studies indicate the usefulness of deep sequencing assay approach to identify both known viruses and new viruses from infected fruit crops. Thus, deep sequencing assay can be used effectively as a first step to control virus diseases in certification programs aimed at elimination of both known and unknown pathogens from plant materials.
Virus-like symptoms of small leaves and/or growth retardation were observed in domestic commercial apple orchards but the disease etiology was unknown. The aim of this study was to apply deep sequencing assays to discover any novel viral genomes and to evaluate the viruses present in apple samples collected in Korea.
Materials and Methods
Plant material and cDNA library construction
Eight samples were sourced from apple trees showing small leaves and/or growth retardation in commercial orchards of Muju, Bonghwa, Boeun, and Yesan provinces during 2011 (Fig. 1). Leaves were collected from symptomatic trees and subjected to laboratory analysis to define the etiology. Total RNA was extracted using the Tri Reagent (Molecular Research Center, Cincinnati, OH, USA) following manufacturer’s instructions. Equal amounts of total RNA from each sample were pooled. Ribo-Zero Magnetic Kit (Epicentre, Madison, WI, USA) was used to remove ribosomal RNA (rRNA) from the total RNA for cDNA library construction and sequencing. A random-primed cDNA library was constructed using a TrueqRNA sample prep Kit (Illumina, San Diego, CA, USA) and BluePippin 2% Agarose Gel Cassettes (Sage Science, Beverly, MA, USA), targeting fragments ranging between 300 bp and 400 bp.

Characteristic symptoms in apple trees show growth retardation from Muju (A), small leaves and leaf curling from Bonghwa (B), small leaves from Bonghwa (C), growth retardation and leaf chlorosis from Bonghwa (D), growth retardation from Yesan (E), small leaves and growth retardation from Yesan (F), small leaves and shortened internodes from Bonghwa (G), and small leaves and leaf curling from Boeun (H).
Sequencing and sequence analysis
The library was sequenced on an Illumina HiSeq 2500 to generate 101 nucleotide (nt) paired-end reads. The paired-end sequence reads were filtered by the NGS QC Toolkit v2.3.3 (Patel and Jain, 2012) to eliminate low quality sequences (phred score ≤ 25) and short reads (length ≤ 20). In addition, the filtered raw sequence reads mapped to the known plant mRNA and the other RNA collections derived from PlantGDB were discarded. De novo assemblies were performed with Velvet version 1.2.09 (Zerbino and Birney, 2008) and the assembled contigs were subjected to search for viral genome sequences using BLAST version 2.2.26. A comparative taxonomic analysis of the BLAST results was performed with MEGA version 5 (Tamura, et al., 2011).
RT-PCR and phylogenetic analysis
Partial genomes of five viruses were retrieved from the BLAST results of assembled contigs as follows: ACLSV, ASGV, ASPV, Apple green crinkle associated virus (AGCaV), and ApLV. RT-PCR was used to confirm the presence of five viruses in the eight symptomatic apple samples. Primers were designed according to the sequences of each assembled contig related to the five viral genomes identified (Table 1). For phylogenetic analysis, nucleic acid sequences were aligned using Clustal W and trees were generated with MEGA v5.05 program (Tamura et al., 2011) using neighbor-joining method.
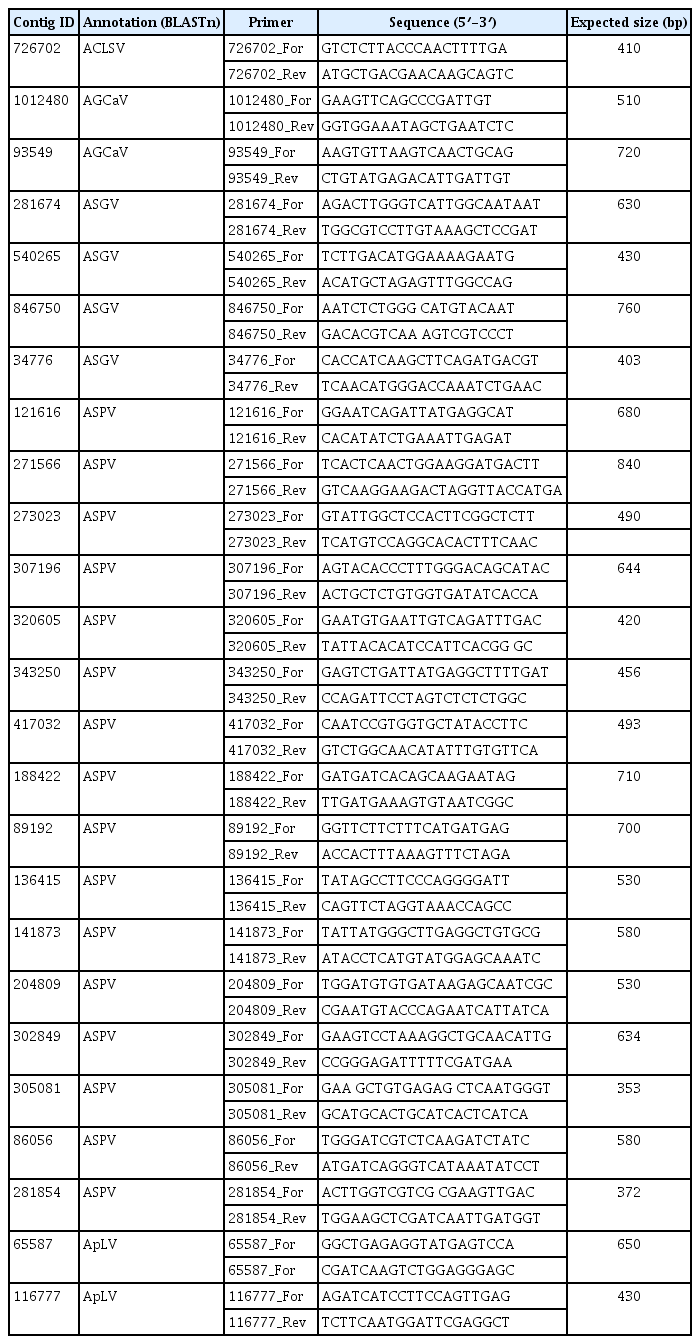
List of the primers used to detect assembled sequences of five viral genomes
Results
Deep sequencing analysis
The deep sequencing of eight apple samples produced paired-end sequence data of 486,469,736 reads. After plant sequence filtering for non-viral RNAs, 319,249,571 raw reads were obtained and subjected to de novo assembly, generating a total of 1,288,953 contigs. BLAST search of the assembled contigs identified fifty two contigs ranging from 500 to 3,300 nt in size with an average of 900 nt in the reference sequences of National Center for Biotechnology Information virus database, which suggested significant similarity with five viruses: ACLSV, AGCaV, ASGV, ASPV, and ApLV (Table 2). These viruses all belong to the familyBetaflexiviridae, i.e., genus Trichovirus (ACLSV), Foveavirus (AGCaV, ASPV, and ApLV), and Capillovirus (ASGV). The nt sequence comparison of the assembled contigs showed from 68% to 99% identity compared to the reference sequences of the five known viral genomes. ASPV and ASGV were the most abundant virus species in this census. ASPV constituted 51.9%, ASGV 34.6%, ApLV 5.8%, ACLSV 3.9%, and AGCaV 3.9% of the fifty two contigs (Fig. 2). ApLV and AGCaV were not previously reported in domestic apple commercial orchards, and were thus identified here for the first time in Korea.
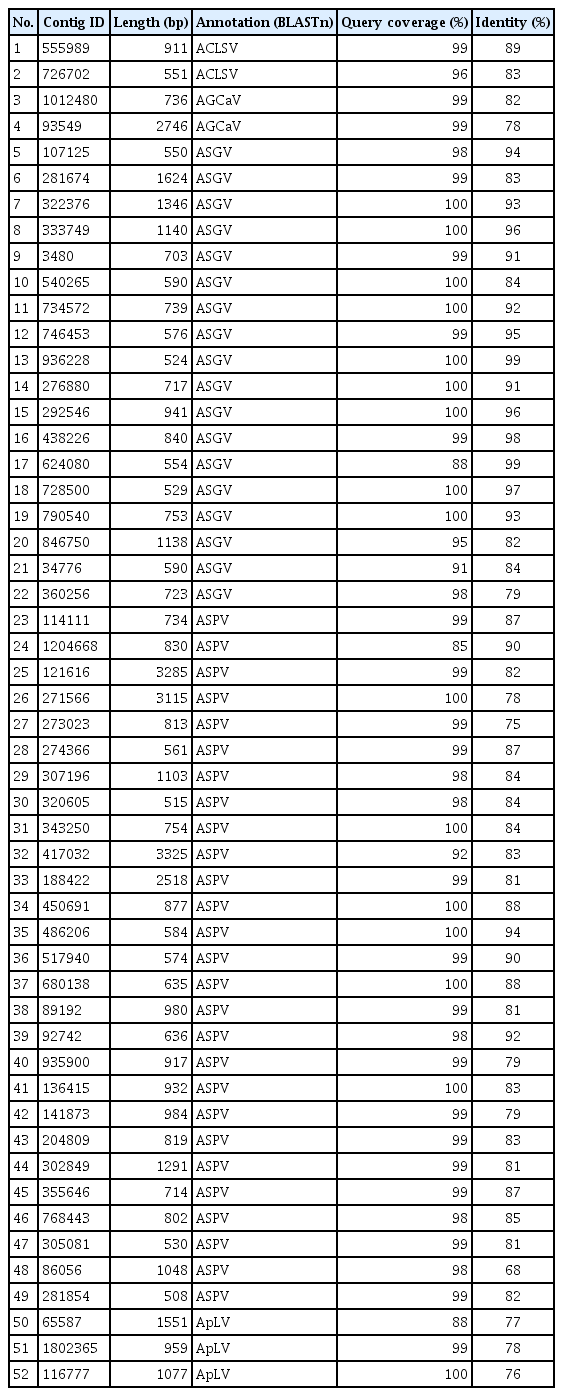
List of fifty two contigs for viral genomes identified according to alignment in the reference sequences of National Center for Biotechnology Information virus database with BLAST searches
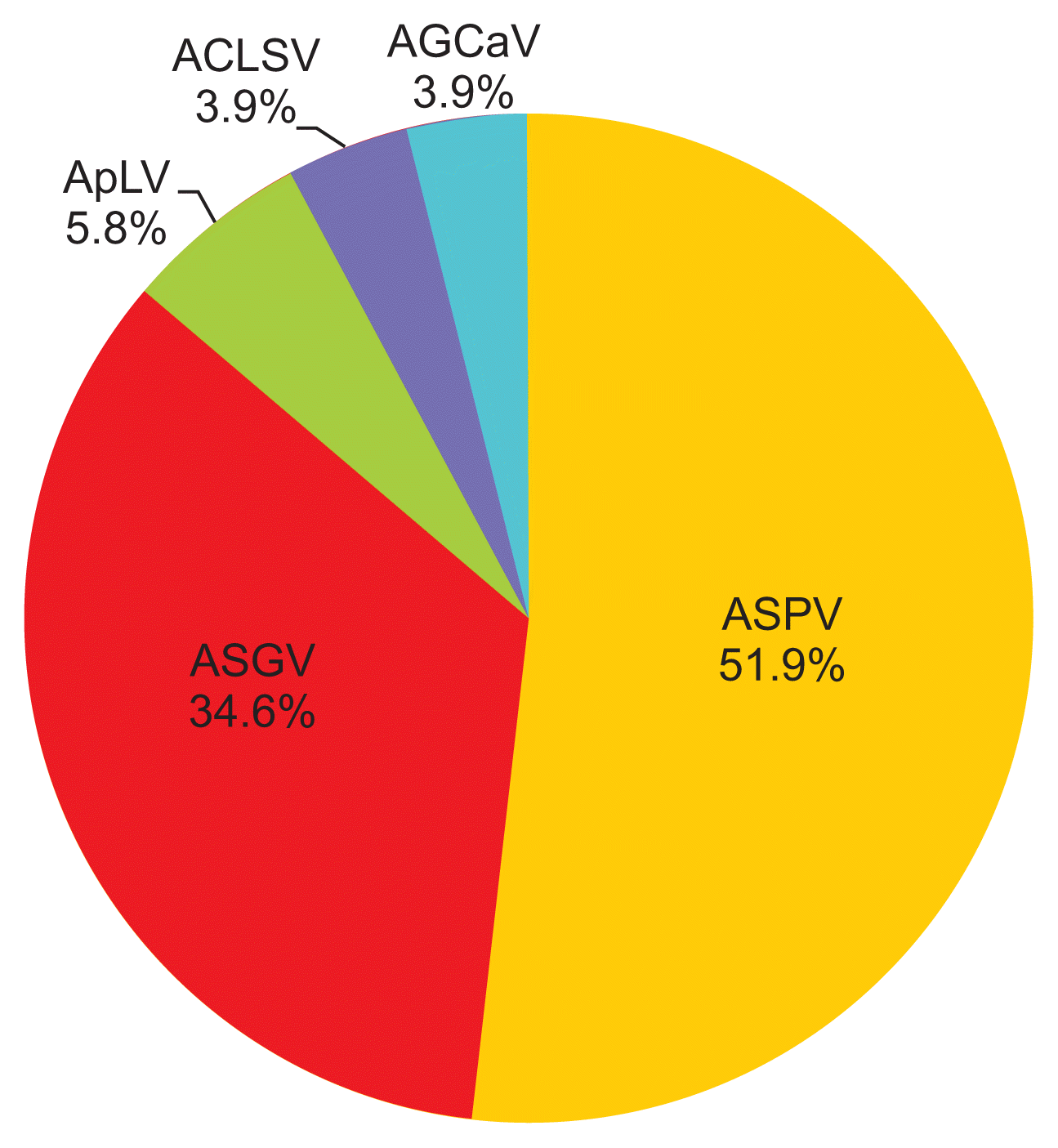
Comparative percentages of five viral genomes among fifty two contigs according to alignment with BLAST searches. ASPV, Apple stem pitting virus; ASGV, Apple stem grooving virus; ApLV, Apricot latent virus; ACLSV, Apple chlorotic leaf spot virus; AGCaV, Apple green crinkle associated virus.
Some of the ASGV contigs were initially identified by BLAST search as most closely related to database sequences labeled as ‘Citrus tatter leaf virus’ (CTLV), which was initially described as a separate species affecting distinct hosts. However, ‘CTLV’ has been recognized by the International Committee on Taxonomy of Viruses (ICTV) since the seventh report (Martelli et al., 2000) as a strain of ASGV. We therefore refer here only to ASGV, although overlapping contigs with distinct sequences were identified for several regions of the ASGV genome (Fig. 3C).

Position and distribution of the assembled contigs in five viral genomes: (A) ACLSV, (B) AGCaV, (C) ASGV, (D) ASPV, and (E) ApLV. MET, methyltransferase; P, papain-like protease; HEL, nucleotide triphosphate-binding helicase; POL, RNA-dependent RNA polymerase; MP, movement protein; CP, coat protein; C, cysteine protease; V, variable region.
Detection of the five viruses in the samples
The presence of five viruses was confirmed by RT-PCR from the eight apple tree samples. All contig sequences derived from deep sequencing were represented on the viral genomes of ACLSV, ASGV, ASPV, AGCaV, and ApLV, respectively (Fig. 3) and primers were designed based on the sequences of each assembled contig (Table 1).
In RT-PCR using the designed primers, the five viruses were successfully detected in the samples as shown in Table 3. All five viruses were detected in one sample from Bonghwa and two from Yesan, all showing small leaves and/or growth retardation; however, only ASGV and ASPV were detected in a sample from Muju with the same growth retardation symptoms as observed in Yesan. These results suggested that the relationship of these viruses to symptom expression in apple trees was not clear. Two or three viruses were detected in each of the other samples. The most frequently detected virus was ASPV, followed by ASGV, ApLV, ACLSV, and AGCaV, which were each detected in multiple infections in the samples tested; none of the samples were infected by less than two viruses (Table 3). Although AGCaV and ApLV were identified here for the first time in Korea, each was present in five to six samples from Bonghwa, Yesan, and Boeun, which indicated that these viruses may occur at significant frequency in domestic commercial apple orchards.

Viruses of eight apple samples detected by RT-PCR
Sequence analysis of newly identified viruses
AGCaV was identified in two contigs, ID 1012480 and 93549. These two contigs were respectively 736 and 2,746 nt in length, showing 82% and 78% nt sequence identity with ORF1 of the AGCaV genome retrieved from GenBank (accession No. HE963831). The phylogenetic relationship between the AGCaV isolate identified from these two contigs and viruses in the genus Foveavirus such as ASPV, ApLV, PCMV, Asian prunus virus (APV), and Grapevine rupestris stem pitting virus (GRSPV) was investigated because it is known that members of the genus Foveavirus display high levels of genetic diversity (James et al., 2013; Komorowska et al., 2011; Youssef et al., 2011). In phylogenetic analysis, the contigs ID 93549 and 1012480 were most closely related to AGCaV than to ASPV and ApLV, respectively, but had a more distant relationship to APV and GRSPV (Fig. 4). The results suggested that the contig ID 93549 and 1012480 might represent new isolates of AGCaV. The contig ID 1012480 of AGCaV isolate was detected in samples B–F and H of Fig. 1 (Fig. 5). The contig ID 93549 of AGCaV isolate was detected only in samples D and H of Fig. 1 (Fig. 5).

Phylogenetic analysis of the genome sequence showing the relationship of contig ID 1012480 (A) and 93549 (B) of AGCaV isolates with available sequences in the genus Foveavirus. Viruses include ASPV, ApLV, PCMV, APV, and GRSPV. AGCaV, Apple green crinkle associated virus; ASPV, Apple stem pitting virus; ApLV, Apricot latent virus; APV, Asian prunus virus; PCMV, Peach chlorotic mottle virus; GRSPV, Grapevine rupestris stem pitting virus.
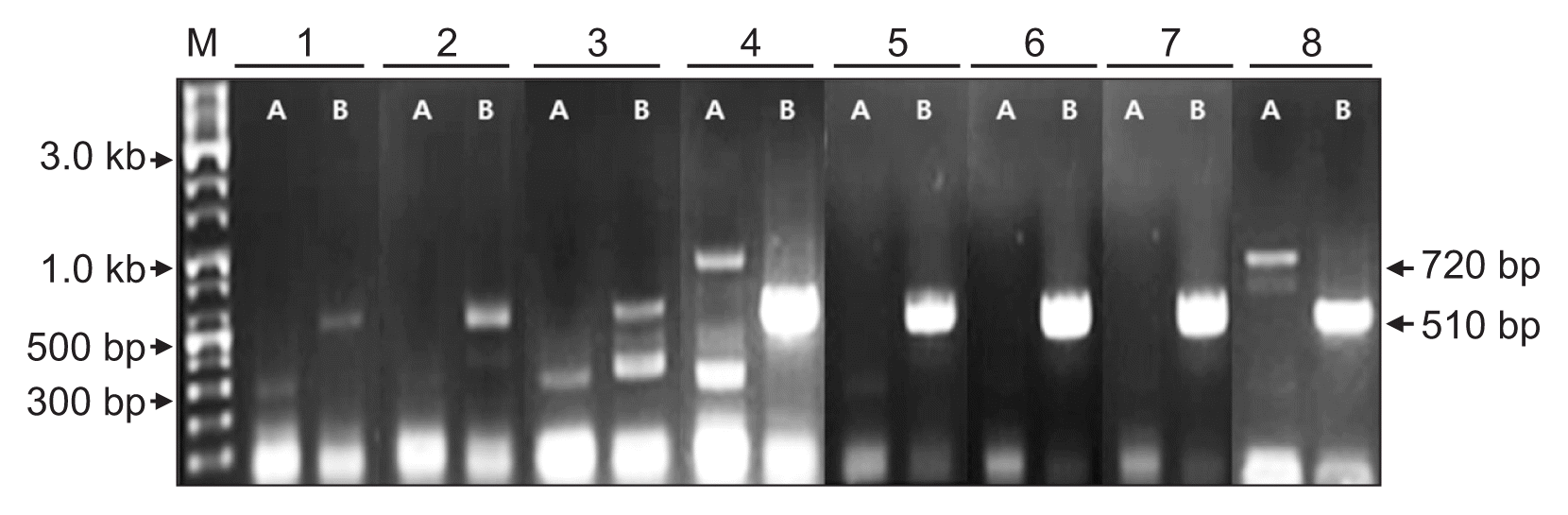
Confirmation of the presence of Apple green crinkle associated virus isolates in the infected apple samples by RT-PCR using primers derived from contig ID 93549 (A) and 1012480 (B). Lanes 1 to 8 correspond to samples A to H of Fig. 1. M, 10 kb ladder DNA marker. Arrows indicate the size of the specific PCR products.
ApLV was identified in three contigs, ID 65587, 1802365, and 116777. These three contigs of 1,551, 959, and 1,027 nt in length showed 77%, 78%, and 76% nt sequence identity respectively with ORF1 of ApLV genome retrieved from GenBank (accession No. HQ339958). In phylogenetic analysis of viruses in the genus Foveavirus, the contig ID 116777 was most closely related to ApLV but only distantly to the type isolates of APV and GRSPV (Fig. 6). The contigs ID 65587 and 1802365 had similar relationships (data not shown). The results suggested that the three contigs might represent new isolates of ApLV. The contig ID 116777 of ApLV isolate was detected in samples D–F and H of Fig. 1 (Fig. 7).
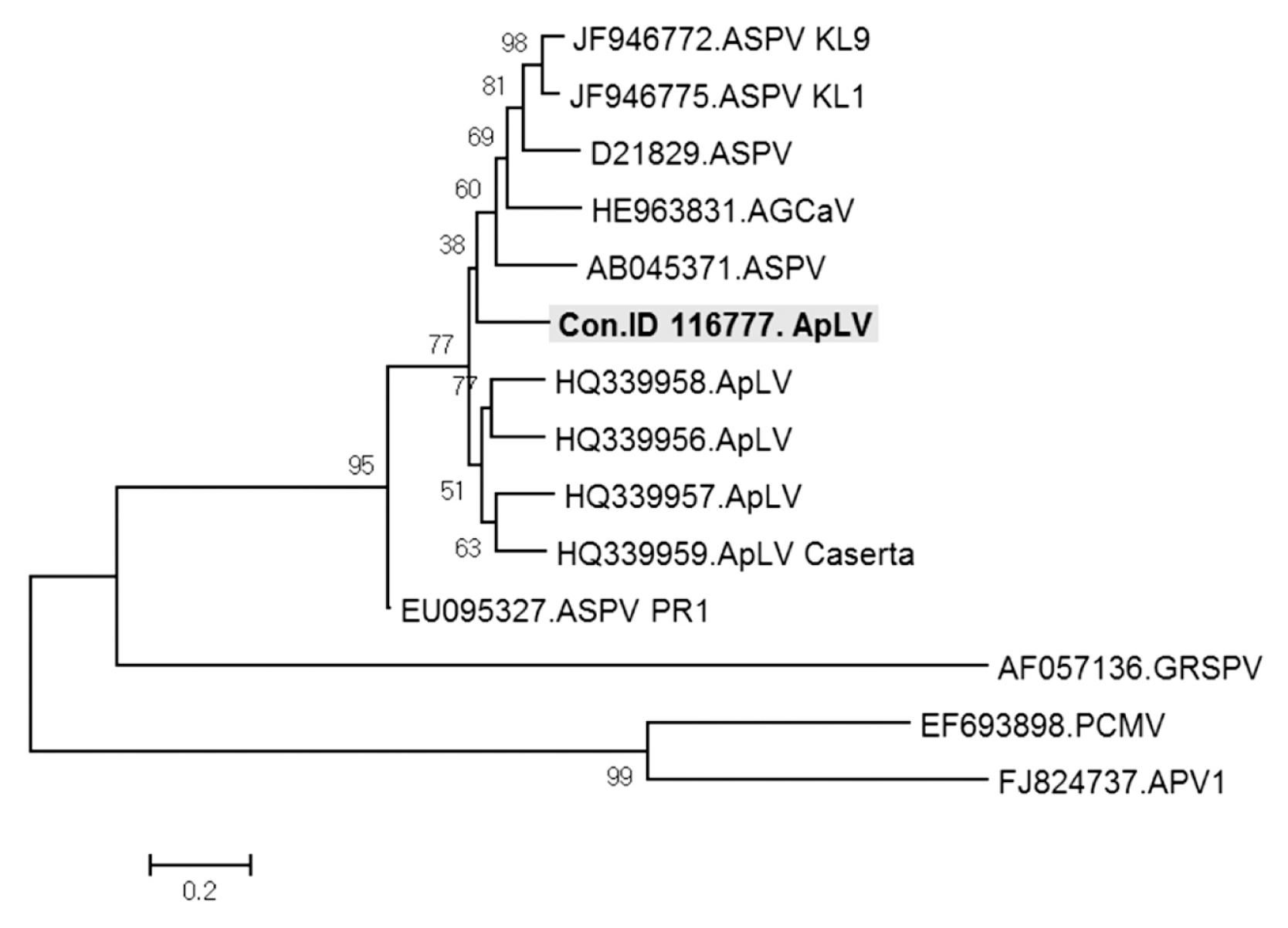
Phylogenetic analysis of the genome sequence showing the relationship of contig ID 116777 ApLV isolate to available sequences in the genus Foveavirus. ASPV, Apple stem pitting virus; AGCaV, Apple green crinkle associated virus; ApLV, Apricot latent virus; GRSPV, Grapevine rupestris stem pitting virus; PCMV, Peach chlorotic mottle virus; APV, Asian prunus virus.

ASPV was identified in twenty-seven contigs, ranging from 508 to 3,325 nt in size. These contigs had DNA sequences with from 68% to 94% identities to known ASPV isolates. Especially, contig ID 86056 of 1,048 nt in length showed only 68% nt sequence identity with ORF1 of ASPV genome retrieved from GenBank (accession No. KF915809). In phylogenetic analysis, this contig was related to ASPV, but appeared to be a distinct virus species of the genus Foveavirus (Fig. 8). The results suggested that the contig ID 86056 is derived from a new Foveavirus most closely related to ASPV. Presence of the sequence represented by contig ID 86056 was confirmed in samples D and H of Fig. 1 by RT-PCR with specific primers (Fig. 9).
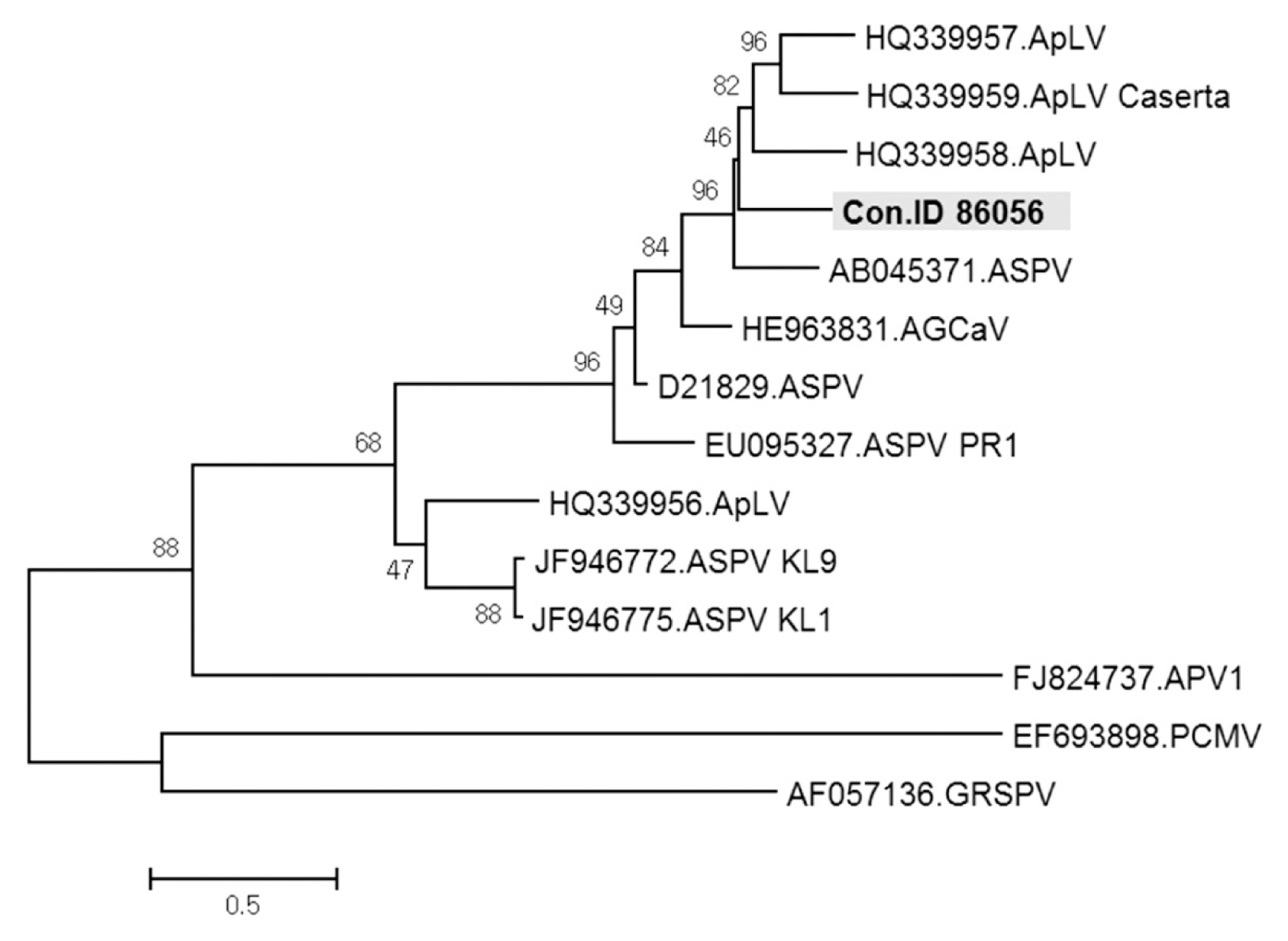
Phylogenetic analysis of the genome sequence showing the relationship of Contig ID 86056 to available sequences of viruses in the genus Foveavirus. ApLV, Apricot latent virus; ASPV, Apple stem pitting virus; AGCaV, Apple green crinkle associated virus; APV, Asian prunus virus; PCMV, Peach chlorotic mottle virus; GRSPV, Grapevine rupestris stem pitting virus.
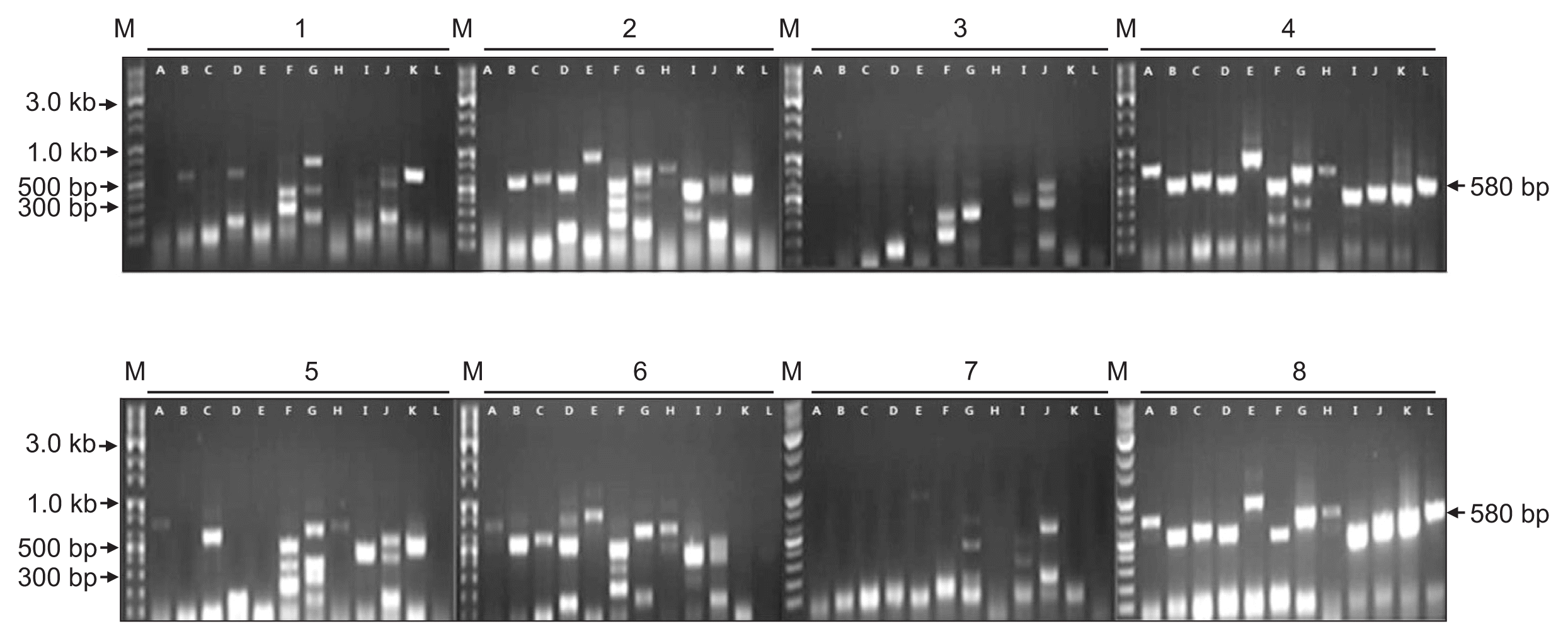
Confirmation of the presence of Apple stem pitting virus in infected apple samples by RT-PCR using primers derived from contig ID 86056. Numbers 1 to 8 correspond to samples A to H of Fig. 1. Lane A, contig ID 89192; lane B, contig ID 136415; lane C, contig ID 141873; lane D, contig ID 204809; lane E, contig ID 271566; lane F, contig ID 273023; lane G, contig ID 302849; lane H, contig ID 307196; lane I, contig ID 320605; lane J, contig ID 343250; lane K, contig ID 417032; lane L, contig ID 86056. M, 10 kb ladder DNA marker. Arrows indicate the size of the specific PCR product of contig ID 86056.
Discussion
There are over 25 diseases of unknown etiology that affect apple trees (Howell et al., 2011). Recently, deep sequencing assay has revealed sequences associated with some diseases of unknown etiology (Barba et al., 2014). In this study, the deep sequencing has generated fifty two contigs related to five viruses, ACLSV, ASGV, ASPV, AGCaV, and ApLV from eight apple samples showing small leaves and/or growth retardation. The presence of five viruses was confirmed in the samples by RT-PCR using specific primers based on the sequences of each of the selected contigs for each virus.
The ASPV isolates were the most abundant virus species in the assembled fifty two contigs, having sequence variants with from 68% to 94% identities with previously reported ASPV isolates. In previous studies, the variable regions of ASPV genome are located between the MET and P-Pro domains, and located in the putative movement protein overlapping the N-terminal coat protein (CP) coding region in pome fruit trees (Gadiou et al., 2010; Magome et al., 1999; Yoshikawa and Takahashi, 1988). Especially, the contig ID 86056 detected in only two samples had a nt sequence that was 68% identical when compared to the corresponding sequence of the definitive ASPV isolate YT. Based on the ICTV criteria for species demarcation in the genus Foveavirus, distinct species have less than ca. 72% identical nt or 80% identical aa between their entire CP or replication protein genes (Adams et al., 2004). The contig ID 86056 might represent a new Foveavirus species. Although the result of the genetic diversity of ASPV isolates in this work is very similar with previous reports, the ASPV complete genomes of the assemble contigs need further study to improve understanding of the extent of genetic diversity.
The detection of ‘ASGV-like’ and ‘CTLV-like’ sequences among the ASGV contigs is similar to the report of Yoshikawa et al. (1996), in which a typical ASGV isolate and a CTLV-like isolate were detected in a mixed infection in Japanese pear. In this study both typical ASGV and a ‘CTLV-like’ isolate were detected in a sample from Yesan, suggesting that mixed infection of a single tree by two distinct isolates is probably a commonplace occurrence for ASGV, as has been shown for several other viruses including the potexviruses Pepino mosaic virus (PepMV; Maroon-Lango et al., 2005), Alternanthera mosaic virus (AltMV; Lim et al., 2010), and Plantago asiatica mosaic virus (PlAMV; Komatsu et al., 2008).
AGCaV and ApLV identified from the assembled contigs were not previously known or studied in Korea. AGCaV was identified in two contigs assembled of contig ID 1012480 and 93549, which showed 82% and 78% nt sequence identities with ORF1 of AGCaV isolate Aurora-1. AGCaV was identified from Aurora Golden Gala apple showing severe symptoms of green crinkle disease (James et al., 2013). Although AGCaV amplicons derived from contig ID 101240 were detected in six of the samples (B–F and H), the green crinkle symptom of apples could not be observed during the survey. Sequences related to AGCaV contig ID 93549 were detected in only two samples (D and H), suggesting that at least two AGCaV variants are present among the trees sampled; this indicates either the probability of multiple introductions into Korea, or of the presence and divergence of AGCaV over a long period. ApLV was identified in three contigs assembled of contig ID 65587, 1802365, and 116777, which showed 77%, 78%, and 76% nt sequence identities with ORF1 of ApLV isolate LA2. Although ApLV might not have been considered fully distinct at the time of the report of Nemchinov et al. (2000), it has been recognized as a species separate from ASPV by the ICTV since 2004. ApLV is mostly latent in the natural host such as peach and cherry, but not in apple plants (Németh, 1986). Although ApLV isolates were detected in five samples by RT-PCR, the analysis of complete genome is needed for the accurate identification of virus species in the genus Foveavirus.
Failure to detect ApMV is consistent with absence of detection of ApMV in other surveys of fruit tree viruses in Korea (Cho, 2015). Although all of the other viruses detected are flexuous viruses with poly-adenylated genomes, the cDNA libraries used for deep sequencing were prepared using random primers, which should have resulted in detection of ApMV if it had been present in any of the samples tested.
These results show that the deep sequencing assay was a significant and powerful tool for detection and identification of known and unknown viruses in the virome of infected apple trees. Accordingly, the assay can be applied to the other fruit crops such as pear, grapevine, peach, and persimmon for virome discovery, and the identification of viruses associated with diseases of unknown etiology in Korea. Furthermore, the assay will be available for the monitoring of viruses for quarantine and routine diagnosis in certification programs by providing fast and accurate virome information in the near future.
Acknowledgments
This research was carried out with the support of “Cooperative Research Program for Agriculture Science & Technology Development (Project No. PJ01007702)” Rural Development Administration, Republic of Korea.
Notes
Articles can be freely viewed online at www.ppjonline.org.