Investigation of Genetic Diversity of Fusarium oxysporum f. sp. fragariae Using PCR-RFLP
Article information
Abstract
Fusarium wilts of strawberry, caused by Fusarium oxysporum f. sp. fragariae, is a serious soil-borne disease. Fusarium wilt causes dramatic yield losses in commercial strawberry production and it is a very stubborn disease to control. Reliable chemical control of strawberry Fusarium wilt disease is not yet available. Moreover, other well-known F. oxysporum have different genetic information from F. oxysporum f. sp. fragariae. This analysis investigates the genetic diversity of strawberry Fusairum wilt pathogen. In total, 110 pathogens were isolated from three major strawberry production regions, namely Sukok, Hadong, Sancheong in Gyeongnam province in South Korea. The isolates were confirmed using F. oxysporum f. sp. fragariae species-specific primer sets. Polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) analyses were executed using the internal transcribed spacer, intergenic spacer, translation elongation factor1-α, and β-tubulin genes of the pathogens and four restriction enzymes: AluI, HhaI, HinP1I and HpyCH4V. Regarding results, there were diverse patterns in the three gene regions except for the β-tubulin gene region. Correlation analysis of strawberry cultivation region, cultivation method, variety, and phenotype of isolated pathogen, confirmed that genetic diversity depended on the classification of the cultivated region.
Introduction
Strawberries belong to the Rosaceae family and are important cash crops not only in South Korea but worldwide. Strawberry cultivars resistant to pests and pathogens have been developed and many strawberry varieties are cultivated in Korea, for example Janghee, Maehyang, Sulhyang. Most cultivars have good flavor and shape, but they have poor hardness, except Maehyang. For this reason, cv. Maehyang has been cultivated as an exportable variety, but it is difficult to cultivate due to its sensitivity to diseases (Park et al., 2012). Strawberry production and exports are on the rise worldwide including South Korea, which is the sixth largest strawberry production country and in terms of exports of strawberries is ranked 18th (Kim et al., 2015).
Strawberries can suffer from various diseases, these typically being bacterial angular leaf spot, anthracnose, gray mold, powdery mildew and Fusarium wilt (Mass, 2013). Fusarium wilt is caused by a soil-borne pathogen, blocking the crown, resulting in leaf anomalies, and the leaves are rolled and browned. It is difficult to control by soil-borne pathogens and lack of information on causative strains (Cha et al., 2016; Fang et al., 2011; Katan, 1999; Koike et al., 2009; Matuo et al., 1980; Nelson, 1981).
Strawberry Fusarium wilt is caused by the soil fungi F. oxysporum f. sp. fragariae, which also serves as a chemical but is difficult to control due to the buffering action of the soil (Couteaudier and Alabouvette, 1990; Nelson, 1981). In addition, mycological characteristics of pathogens also make control difficult (Katan et al., 1997; Rekah et al., 2000). F. oxysporum f. sp. fragariae consists of three types of spore, of which the microconidia are the smallest, followed by the macroconidia, which are larger in size and known to cause disease. The largest is the chlamydospore which is resistant to adverse environmental conditions. Chlamydospores can remain dormant in soil for as long as 30 years, and all of these spores can spread through running water, farm machinery and tools (Couteaudier and Alabouvette, 1990; Koike et al., 2009; Nelson, 1981). Unlike other well-known strains, F. oxysporum f. sp. fragariae is known to cause disease only in strawberry (Koike et al., 2009; Mass, 2013), while other strains belonging to Fusarium spp. have been studied and reported for their genetic information. However, information on this particular F. oxysporum f. sp. fragariae strain is very limited (Armstrong and Armstrong, 1981; Edel et al., 1995).
In general, sequencing of the internal transcribed spacer (ITS) is used for the classification and identification of fungi (White et al., 1990). However, it is insufficient to identify complex and variable genes and therefore, cannot confirm any genetic variation. Unlikely with the genus Fusarium, the genetic information for F. oxysporum f. sp. fragariae has not been reported. However, information on pathogen genetic diversity and variation is necessary to understand pathology and development control methods. Molecular markers have been used to identify systematic relationships and diversity between pathogens and diseases. Of the various technology that has been developed, the restriction fragment length polymorphism (RFLP) pattern of rDNA was employed to confirm genetic variation and diversity of plant pathogenic fungi. RFLPs are useful for grouping isolated strains at less cost without the need for expensive equipment and experiments (Bogale et al., 2007; Hausner and Wang, 2005; Hillis and Dixon, 1991; Kachuei et al., 2015; Manicom et al., 1990).
Fusarium wilt causes severe damage in strawberry production. Therefore, this study was conducted to: firstly, analyze the genetic variation and diversity of F. oxysporum f. sp. fragariae, which causes Fusarium wilt; and secondly, construct mycological and genetic information of the pathogen.
Materials and Methods
Strawberry field and sampling site description
In 2015 and 2016, three major strawberry cultivation regions in Gyeongnam province were investigated for Fusarium wilt disease symptoms. The main strawberry production areas (Sukok, Hadong, and Sancheong) used well established soil cultivation systems for cultivars such as Jangee, Sulhyang, Maehyang, Santa, and Amaka. During the cultivation season, Fusarium wilt disease symptoms were documented, including irregular leaf development, yellowish leaf and decay in the crown part of the plant. The Fusarium wilt infected plants were collected from the sites and systematically identified.
Isolation of Fusarium wilt pathogens
The crown parts with disease lesions were cut (0.5 × 0.5 cm) and their surface sterilized by 1% sodium hypochlorite for 30 s and 70% ethanol for 30 s. The segments were continuously washed 5 times with sterilized water then dried for 10 min under clean bench, placed on water agar medium (WA; agar 20 g per 1 l) for 3 days at 28°C. The grown hypha was transferred to a potato dextrose agar (PDA; potato dextrose 24 g, agar 20 g per 1 l) for 5 days at 28°C. From the three cultivation regions of Sukok, Hadong, and Sancheong, a total of 110 pathogens were isolated as the symptoms.
Identification of the isolates’ morphological and molecular characteristics
The isolates were transferred from WA to PDA and then incubated for 5 days at 28°C. The contamination was observed for 5 days and the mycelium color of the uncontaminated isolates was observed. The isolates having the same color were collected and classified as different phenotype groups. Prior to preparing spore suspensions of the pathogen hypha classified by color, the spore suspensions were poured into 10 ml of sterilized water into a mold-cultured plate. Optical microscope (Carl Zeiss A.G., Oberkochen, Germany) observations (×400) were performed to determine the spores’ type and morphology and a 50 μm meter scale bar was used.
Total genomic DNA was extracted at the mycelium using cetyl trimethyl ammonium bromide (CTAB) protocol (Porebski et al., 1997). The extracted DNA from each isolate was amplified with F. oxysporum f. sp. fragariae specific primers; FofraF and Fofra R (Suga et al., 2013). The PCR conditions were as follows: initial denaturation at 94°C for 2 min, followed by 30 cycles of 94°C for 30 s, 55°C for 30 s, and at 72°C for 15 s. The final extension temperature was set at 72°C for 5 min. The complete ITS region of ribosomal RNA was amplified with the primers ITS1 and ITS4 (Kistler et al., 1987; White et al., 1990) then sequencing was done using Solgent Co. (Daejeon, Korea). Sequencing similarities were compared to previously deposited sequences in NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
PCR-RFLP and phylogenetic analysis
ITS, intergenic spacer (IGS), translation elongation factor1-α (TEF-1α), and β-tubulin were employed for PCR-RFLP analysis. PCR was used with PCR PreMix (Bioneer, Daejeon, Korea) including 10 pmol of the primer sets and 50 ng of fungal genomic DNA. ITS1 and ITS4 (Kistler et al., 1987; White et al., 1990) for ITS and PCR conditions were as follows: initial denaturation at 95°C for 5 min, followed by 30 cycles of 95°C for 1 min, 50°C for 1 min, and at 72°C for 1 min and the final extension temperature was set at 72°C for 10 min for ITS. To amplify IGS, FIGS and FIGS2 primers were used and the PCR conditions were as follows: initial denaturation at 95°C for 5 min, followed by 30 cycles of 95°C for 40 s, 52°C for 40 s, and at 72°C for 40 s and the final extension temperature was set at 72°C for 10 min. Primers, TEF fusarium F and fusarium R, and PCR condition was following settings; initial denaturation at 95°C for 5 min, followed by 35 cycles of 95°C for 1 min, 58°C for 1 min, and at 72°C for 2 min and the final extension temperate was at 72°C for 10 min for TEF-1α. For β-tubulin gene, primers FBETA F and FBETA R was used (Table 1) and PCR condition was following settings; initial denaturation at 94°C for 3 min, followed by 35 cycles of 94°C for 40 s, 62°C for 40 s, and at 72°C for 30 s and the final extension temperate was at 72°C for 10 min. The amplified PCR products were subjected to enzymatic cut with the restriction enzymes AluI, HhaI, HinP1I, and HpyCH4V (NEB, Hitchin, UK) according to protocol in 20 μl total volume. The restriction reaction mixtures were incubated at 37°C for 4 h. To remove the enzyme activity continuously the reaction tubes were incubated at 70°C for 15 min. The DNA fragments were then loaded on 3% agarose gel at 120 V or 200 mA for 40 min in 1× TAE buffer. The gels were stained with ethidium bromide (EtBr) for visualization. The product fragments were formulated as ‘0’ or ‘1’. The 0 signified absence while 1 signified the presence of a particular band. Based on PCR-RFLP analysis, unweighted pair group method with arithmetic average (UPGMA) method dendrograms were determined via genetic variance between the isolates and then discriminant analysis and construction according to linear combinations of the variables was used to determine the maximum separation between the isolates using XLSTAT program (www.xlstat.com) (Gang et al., 2015).
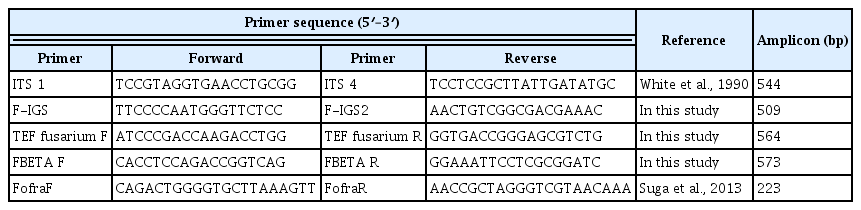
Primers used in this study
Results
Origin and isolation of the Fusarium wilt pathogens
Fusarium wilt diseased strawberries were collected in the Gyeongnam region during the strawberry cultivation period in 2015. The samples were collected from three locales—Sukok, Hadong, and Sancheong and the cultivated types were either high cultivation or soil cultivation. Strawberry cultivars were varieties including Sulhyang, Maehyang, Janghee, Santa, and Amaka. The infected plant showed typical Fusarium wilt disease symptoms (Supplementary Fig. 1). As a result, 38 isolates were isolated from Sukok, 29 from Hadong, and 43 from Sancheong (Supplementary Table 1). When they were classified into cultivated form, 36 isolates were cultivated in high cultivation and 74 isolates were cultivated in soil cultivation. When they were classified based on cultivar, 13 isolates from Maehyang, 67 isolates from Sulhyang, 24 isolates from Janghee, and 3 isolates were isolated from Santa and Amaka (Table 2).

Information on isolates with cultivation conditions
Identification of phenotype, morphological spores and molecular classification
The isolates were classified into five phenotypes. The criterion for classification was the mycelium color of isolated pathogens. The 110 isolated pathogens were classified into 5 different hypha phenotypes and the colors of hyphae were as follows: dark red, light violet, light yellow, white, and dark pink (Fig. 1). The spores of the isolates were classified based on morphological characteristics. Microconidia and macroconidia of the same shape and size as those of F. oxysporum f. sp. fragariae were confirmed. Generally, microconidia have 5–12 × 2.3–3.5 μm and macroconidia have 23–54 × 3–4.5 μm (Supplementary Fig. 2). Furthermore, the results of ITS sequencing for all isolates showed 99–100% similarity with standard F. oxysporum f. sp. fragariae strain (Supplementary Fig. 3).

Classification of isolates by phenotype. (A) Dark red. (B) Light violet. (C) Light yellow. (D) White. (E) Dark pink. The isolates were grown on potato dextrose agar for 5 days at 28°C.
Identification of the pathogen isolates with species-specific PCR
The species-specific primer sets were used to identify the isolated pathogens, Fofra F and Fofra R for F. oxysporum f. sp. fragariae (Suga et al., 2013). PCR results showed a single amplicon (233 bp) (Supplementary Fig. 4) from each region’s agriculture land and confirmed 38 isolates from Sukok, 30 isolates from Hadong and 42 isolates from Sancheong. In total 110 isolated pathogens were identified as F. oxysporum f. sp. fragariae.
PCR-RFLP and phylogenetic analysis
A total of 110 isolates were analyzed by PCR-RFLP for evidence of genetic variation. When the restriction enzyme was applied to the gene amplified from the ITS region, various PCR bands were obtained (Fig. 2). AluI-ITS generated 4 types of diversity pattern of the pathogen. Furthermore HhaI-ITS showed 7 types of diversity pattern, HinP1-ITS indicated 6 types of diversity pattern and HpyCH4V-ITS revealed 4 types of diversity pattern, respectively (Supplementary Fig. 5). The PCR-RFLP results for the IGS and TEF1-α regions revealed two or three patterns. AluI-IGS, HhaI-IGS, HpyCH4V-IGS generated 3 types of diversity pattern; however, HinP1-IGS indicated only 1 pattern and separated the three groups (Supplementary Fig. 6). AluITEF1-α, HhaI-TEF1-α showed 3 types diversity pattern and HinP1-TEF1-α, HpyCH4V-TEF1-α revealed 2 types diversity pattern (Supplementary Fig. 7). In the β-tubulin region, 2 types of pattern were observed when the AluI restriction enzyme was treated, and all of the remaining patterns showed a uniform pattern (Supplementary Fig. 8). PCR-RFLP patterns were analyzed with the XLSTAT program. When examined, it was confirmed that three of the four genes were differentiated into three groups except for the β-tubulin domain, but the β-tubulin domain could be divided only into two groups (Supplementary Fig. 9). When analyzed, it was divided into five groups. To find a correlation genetic diversity of the pathogen and among the criteria such as strawberry cultivated, cultivated cultivar, cultivated region and phenotype. Only cultivation regions showed a reliable correlation grouping among the criteria (Fig. 3, Table 3). Specifically, 95.2% of samples in Sancheong were found only in group 5 and 76.7% of samples in Hadong were detected in group 4 (Table 3, Supplementary Fig. 10). Principal component analysis (PCA) was conducted to clarify the cause analysis of the genetic classification of the pathogen isolates. Three groups were divided in PCA and the grouping was divided by the cultivation regions (Fig. 4).

The restriction fragment length polymorphism patterns. (A) Illustrated fragmentation of internal transcribed spacer (ITS). (B) Illustrated fragmentation of intergenic spacer (IGS). (C) Illustrated fragmentation of translation elongation factor1-α (TEF1-α). (D) Illustrated fragmentation of β-tubulin.

Phylogenic tree constructed from four genes. Based on PCR-restriction fragment length polymorphism analysis, unweighted pair group method with arithmetic average (UPGMA) method dendrograms were determined via genetic variance between the isolates (25% dissimilarity). Dendrograms were determined genetic variance between the isolates and constructed using XLSTAT program.
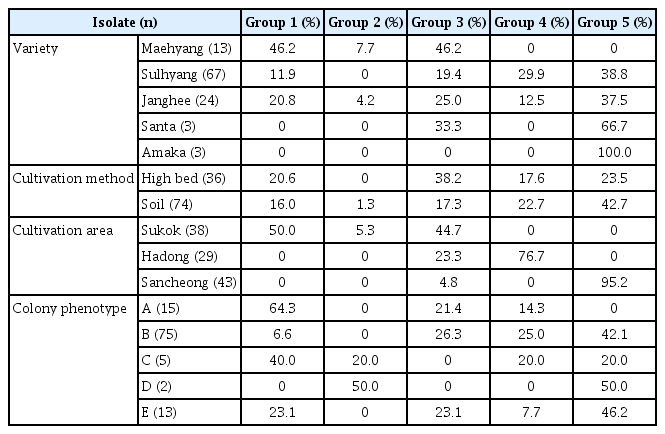
PCR-restriction fragment length polymorphism results showing the percentage of isolates in each group
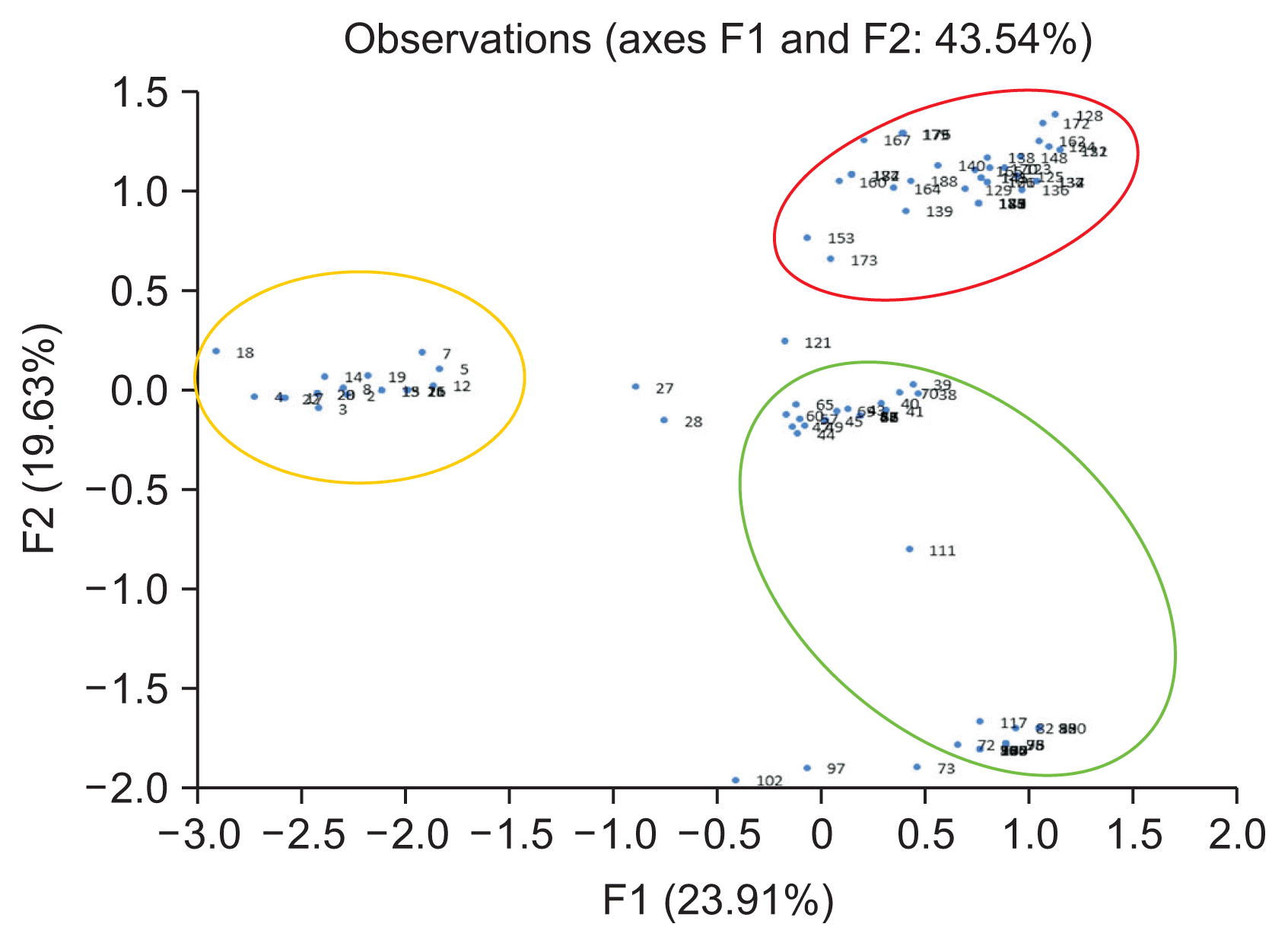
Two-dimensional ordination of 110 the pathogen isolate genotypes on principle component axis 1 and 2. The blue dots and numbers indicate the isolates. The majority of pathogen isolates in red circle were isolated from Sancheong, yellow circle show the Sukok isolates and green circle represent the Hadong isolates. Principle component analysis determined the genetic variance between the isolates and constructed using the XLSTAT program.
Discussion
The purpose of this study is to determine the genetic diversity and variation in the strawberry Fusarium wilt pathogen. It also seeks to generate information on diversity in the pathogen. A total of 110 isolates was confirmed by PCR using F. oxysporum f. sp. fragariae species-specific primers (Suga et al., 2013). The phenotype was divided into 5 phenotypes based on the colony’s morphological characteristics. Despite having different phenotypes, the isolates confirmed that the pathogens were identical to F. oxysporum f. sp. fragariae. In addition, the ITS sequencing of each phenotype demonstrated 99–100% similarity to F. oxysporum f. sp. fragariae. Therefore, in this study the strawberry Fusarium wilt pathogen isolates were consistent with both the molecular identification and morphological characteristics.
In general, the PCR-RFLP analysis is used to study genetic variation and diversity in fungi. While many genetic types of Fusarium spp. have been studied, F. oxysporum f. sp. fragariae has not been reported in terms of its genetic diversity or variation. For PCR-RFLP genetic analysis, ITS, IGS, TEF1, and TUB gene regions are widely used in fungal genetic studies (Donaldson et al., 1995; Dubey et al., 2014; Kim and Min, 2004). In general, the ITS and IGS regions are non-coding region on rDNA and theses gene appears severely variation (Hillis and Dixon, 1991). ITS sequences were proposed as the official barcode markers for fungi (White et al., 1990). In the same way, ITS-RFLP and IGS-RFLP showed a wide variety of patterns, and the ITS-RFLP region had the most patterns among the genes used in this study. In addition, the TEF-1α gene and β-tubulin gene were widely employed in the PCR-RFLP technique because they are easy to use taxonomically. The β-tubulin gene information is often used to study the identification of a pathogen or population (Donaldson et al., 1995; Dubey et al., 2014; Gang et al., 2015; Kim and Min, 2004). However, this study showed that β-tubulin RFLP and phylogenetic analysis Fusarium wilt pathogen is not significantly different from other pathogen isolates. The β-tubulin gene showed only a uniform pattern, and this particular pattern was more varied than expected.
Analysis confirmed that three of the four genes were differentiated into three groups except for the β-tubulin domain, but the β-tubulin domain could be divided only into two groups. When all patterns at were combined at once, it was divided into five groups. It was necessary to find the criteria to classify among cultivar, cultivated region and phenotype. The strawberry cultivation areas are clearly different but there was a correlation between genetic variation and the pathogens. Especially, 95.2% of samples in Sancheong were found only in group 5 and 76.7% of samples in Hadong were found in group 4, respectively. Unlike the other two locales, Sukok isolates were not clearly classified, but 50.0% and 44.7% of the samples were divided into group 1 and group 3. In the comparison based on cultivars, there is a clear classification of cvs. Amaka and Santa, but it is premature make a conclusion due to the sample numbers being relatively small. PCA was conducted to clarify the genetic classification of pathogens and four groups were divided. PCA is a statistical procedure that uses an orthogonal transformation method to convert a set of observations of possibly correlated variables into a set of values of linearly uncorrelated variables. PCA analysis clearly showed that the grouping factors depended on the area of cultivation. The new knowledge generated in this study is that the pathogens’ genetic variation and diversity are largely influenced by where cultivation occurs.
Although the geographical distance between the three strawberry cultivation regions is approximately 30 km, there is a clearly existing genetic variation. Further experiments should be conducted to discover what caused these phenomena in the pathogens. We assume that in the case of Sugok and Hadong, where strawberry cultivation and production has dramatically increased in recent years and the method of cultivation is changing, there is a tendency to purchase seeding plants from different regions. However, in Sancheong, most strawberry farmers are still using self-seeding plants for cultivation. For this reason, it is thought that the isolates from Sancheong are markedly differentiated in terms of pathogen diversity compared to the other two regions. Currently, diversity and genetic information is very limited in F. oxysporum f. sp. fragariae. Therefore, this study is expected to serve as an important groundwork for further genetic variation research on F. oxysporum f. sp. fragariae.
Supplementary Information
Acknowledgments
This study was supported by Technology Development Program for Agriculture and Forestry, Ministry for Food, Agriculture, Forestry and Fisheries, Republic of Korea (Project No. 315004-5).