Occurrence and Evolutionary Analysis of Coat Protein Gene Sequences of Iranian Isolates of Sugarcane mosaic virus
Article information
Abstract
Sugarcane mosaic virus (SCMV) is one of the most damaging viruses infecting sugarcane, maize and some other graminaceous species around the world. To investigate the genetic diversity of SCMV in Iran, the coat protein (CP) gene sequences of 23 SCMV isolates from different hosts were determined. The nucleotide sequence identity among Iranian isolates was more than 96%. They shared nucleotide identities of 75.5–99.9% with those of other SCMV isolates available in GenBank, the highest with the Egyptian isolate EGY7-1 (97.5–99.9%). The results of phylogenetic analysis suggested five divergent evolutionary lineages that did not completely reflect the geographical origin or host plant of the isolates. Population genetic analysis revealed greater between-group than within-group evolutionary divergence values, further supporting the results of the phylogenetic analysis. Our results indicated that natural selection might have contributed to the evolution of isolates belonging to the five identified SCMV groups, with infrequent genetic exchanges occurring between them. Phylogenetic analyses and the estimation of genetic distance indicated that Iranian isolates have low genetic diversity. No recombination was found in the CP cistron of Iranian isolates and the CP gene was under negative selection. These findings provide a comprehensive analysis of the population structure and driving forces for the evolution of SCMV with implications for global exchange of sugarcane germplasm. Gene flow, selection and somehow homologous recombination were found to be the important evolutionary factors shaping the genetic structure of SCMV populations.
Introduction
Mosaic is the most destructive virus disease of sugarcane (Koike and Gillaspie, 1989; Xu et al., 2008). Sugarcane mosaic virus (SCMV; genus Potyvirus, family Potyviridae) is one of the causal pathogens of mosaic diseases on sugarcane, maize, sorghum and some other graminaceous species with worldwide distribution and significant yield losses (Achon et al., 2007; Alegria et al., 2003). Sugarcane originated from the island of New Guinea in the South Pacific and then spread to Northern India. It is now cultivated in several countries in the world, prominently in Brazil, India, China, Pakistan, Mexico, Thailand, Colombia, Australia, Indonesia, Philippines, and USA (He et al., 2016). In Iran, sugarcane is one of the most important commercial crops cultivated in north and southwest of the country. Viral diseases such as those caused by SCMV are a potential threat to sugarcane industry (Moradi et al., 2016). SCMV was first reported in sugarcane from Khuzestan Province of Iran in 1993 (Amiri and Izadpanah, 1993). Like other potyviruses, the virus has a positive single-stranded RNA genome encoding a single polyprotein and is transmitted by aphids in a non-persistent manner. The polyprotein is cleaved by three self-encoded proteinases to produce at least ten final protein products, which are multifunctional (Adams et al., 2012). The coat protein (CP) is a protein involved in several steps of the potyvirus life cycle: aphid transmission, cell-to-cell and systemic movement, encapsidation of the viral RNA, regulation of viral RNA amplification (Urcuqui-Inchima et al., 2001) and probably host specificity (Li et al., 2013; Shukla et al., 1991). The variable N-terminus of the potyvirus CP is necessary for aphid transmission, systemic infection, and adaptation of the virus to its host (Ullah et al., 2003). Differences in this region are important for the potyvirus discrimination (Handley et al., 1998; Shukla and Ward, 1989; Zhong et al., 2005). Evolutionary studies of viruses concentrated on understanding effects of variation caused by mutation, recombination, selection pressure, and host adaptation in viral populations. These studies help us to understand important aspects of viral biology such as changes in virulence, geographical spread and adaptation to new hosts or emergence of a new epidemic (Gibbs and Ohshima, 2010; Li et al., 2013; Moury et al., 2002). Hence, information on evolutionary history of viruses is crucial to design management and control strategies to contain their spread. Non-persistently transmission of SCMV by aphids makes the control of the virus vectors rather inefficient. Therefore, cultivation of resistant varieties is the most promising approach for control of SCMV (Dussle et al., 2002) which in turn requires a complete understanding of the genetic diversity of the virus. Several studies have been performed in recent decades on SCMV biology, genome characterization and sequence diversity (Achon et al., 2007; Alegria et al., 2003; Gao et al., 2011; Wang et al., 2010). Recently, Li et al. (2013) analyzed the genetic diversity and population structure of SCMV based on CP coding region. They suggested that gene flow, recombination and selection pressures were important driving forces for the evolution of SCMV. Based on complete genome sequence analysis, Xie et al. (2016) showed that host, negative selection and recombination were important evolutionary factors shaping the genetic structure of SCMV populations in Shanxi province of China. To our knowledge, little is available on the genetic variability and recombination analysis of the CP gene of SCMV population in Iran. The present study was carried out to investigate the prevalence and genetic diversity of the virus in Khuzestan and Mazandaran provinces of Iran, the major growing regions of sugarcane and maize in the country. To this end, the CP gene sequences of Iranian and worldwide isolates of SCMV were analyzed to know more about the molecular evolution of SCMV populations in Iran.
Materials and Methods
Virus isolates, RNA isolation and RT-PCR
A total of 250 leaf samples of sugarcane (n = 115), maize (n = 96), sorghum (n = 25) and some weeds (Johnson grass = 14) were collected during 2013–2016 growing seasons from two main cultivation regions in Iran, Mazandaran (north) and Khuzestan (south-west) provinces. Collected samples were representatives of 24 randomly selected sugarcane and maize fields, and were of common sugarcane (cvs. CP48-103, CP69-1062, CP57-614, CP63-588, CP78-1628) and maize (cvs. single cross 704, KSC604, KSC647, KSC400, KSC260, KSC703) cultivars. In general, the sugarcane and maize plants were collected in a zig-zag pattern across the field, regardless of the symptoms. Samples were screened for SCMV infection by RT-PCR using SCMV-R3/F4 specific primers (Alegria et al., 2003) to amplify the 900 bp of the CP coding region. Total RNA was extracted from all infected samples using High Pure Viral Nucleic Acid Kit (Roche, Basel, Switzerland) and used as template for reverse transcription test. The first strand cDNA was synthesized using antisense primer and the Moloney murine leukemia virus (MMuLV) reverse transcriptase (Thermo Scientific, Waltham, MA, USA). For this, 4 μl of purified RNA were mixed with 2 μl of antisense primer (10 pmol/μl), heated at 65°C for 5 min and chilled on ice. The mixture was added to reverse transcription mix (50 mM Tris-HCl, pH 8.3, 50 mM KCl, 4 mM MgCl2, 10 mM dithiothreitol, 1 mM of each dNTP, 200 units of MMuLV-RT) in a final volume of 20 μl and incubated at 42°C for 60 min. PCR was carried out using Taq PCR Master Mix (Ampliqon, Odense, Denmark) in a total volume of 25 μl as follows; 12.5 μl Master Mix, 1 μl forward primer (10 pmol/μl), 1 μl reverse primer (10 pmol/μl) followed by 7.5 μl nuclease-free water and finally 3 μl of cDNA. The PCR thermal profile was 95°C for 3 min; followed by 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 90 s; and 72°C for 7 min as a final extension after the last cycle. The amplified fragments were visualized on agarose gel (1%) and from SCMV-positive samples set, 23 specimens were chosen according to hosts and locations. Subsequently, the PCR products of the samples were extracted from the gel and purified with the Qiaquick Gel Extraction Kit (Qiagen, Hilden, Germany).
Cloning and sequencing
The purified PCR products of 23 isolates were ligated into pTZ57R/T vector (Thermo Scientific), according to the manufacturer’s instructions. The recombinant vectors were transformed into competent cells of Escherichia coli DH5α. The positive clones were confirmed by colony-PCR using M13-forward and reverse primers. Plasmid DNA from recombinant clones was purified using a Plasmid Miniprep Kit (Qiagen). The inserts of recombinant plasmids were sequenced in both directions (Macrogen, Seoul, Korea). At least two independent clones were sequenced for each of the amplicons. Sequence data were assembled using the contig Express program in the Vector NTI 11 software (Invitrogen, Carlsbad, CA, USA). Consensus sequences were analyzed using the BLAST program in the National Center for Biotechnology Information database.
Sequence alignment
Nucleotide (nt) and amino acid (aa) sequence alignments of the CP genes of 103 SCMV isolates, including 27 Iranian SCMV isolates (23 isolates of this study plus 4 isolates previously deposited in GenBank including KhzL66 [DQ369960], KhzQ86 [DQ438949], NRA [KT895080] and ZRA [KT895081]) and 76 isolates from other countries, were generated using the DNA-MAN 7 software (Lynnon Biosoft, Vaudreuil-Dorion, QC, Canada) and ClustalW program in BioEdit ver.7.2.5 software (Ibis Biosciences, Carlsbad, CA, USA).
Test of recombination
The aligned CP nucleotide sequences were examined for the presence of recombination events using seven recombination detection methods implemented in the RDP4 software (Martin et al., 2015) with default parameters and the highest acceptable P-value of 0.05. Only potential recombination events detected by at least four different methods, coupled with phylogenetic evidence of recombination, were considered significant.
Phylogenetic analysis
The pairwise nucleotide and amino acid sequence identity scores were represented as color-coded blocks using SDT v.1.2 software (Muhire et al., 2013). Phylogenetic trees for the CP gene of all isolates were constructed by the neighbor-joining (NJ) and maximum likelihood (ML) methods using MEGA v.6 (Tamura et al., 2013) and just the NJ tree is shown (Fig. 1). Maize dwarf mosaic virus-Bg (GenBank accession no. AJ001691) was used as an outgroup because BLAST searches have shown it to be the sequence in the International nucleotide sequence databases most closely and consistently related to the genomic sequence of SCMV. The integrity of the evolutionary relationships was assessed by 1,000 bootstrap replicates. The within- and between-group estimates of evolutionary divergence (d) were calculated for the CP sequences of the 103 SCMV isolates based on the identified SCMV phylogroups, using MEGA v.6 software.
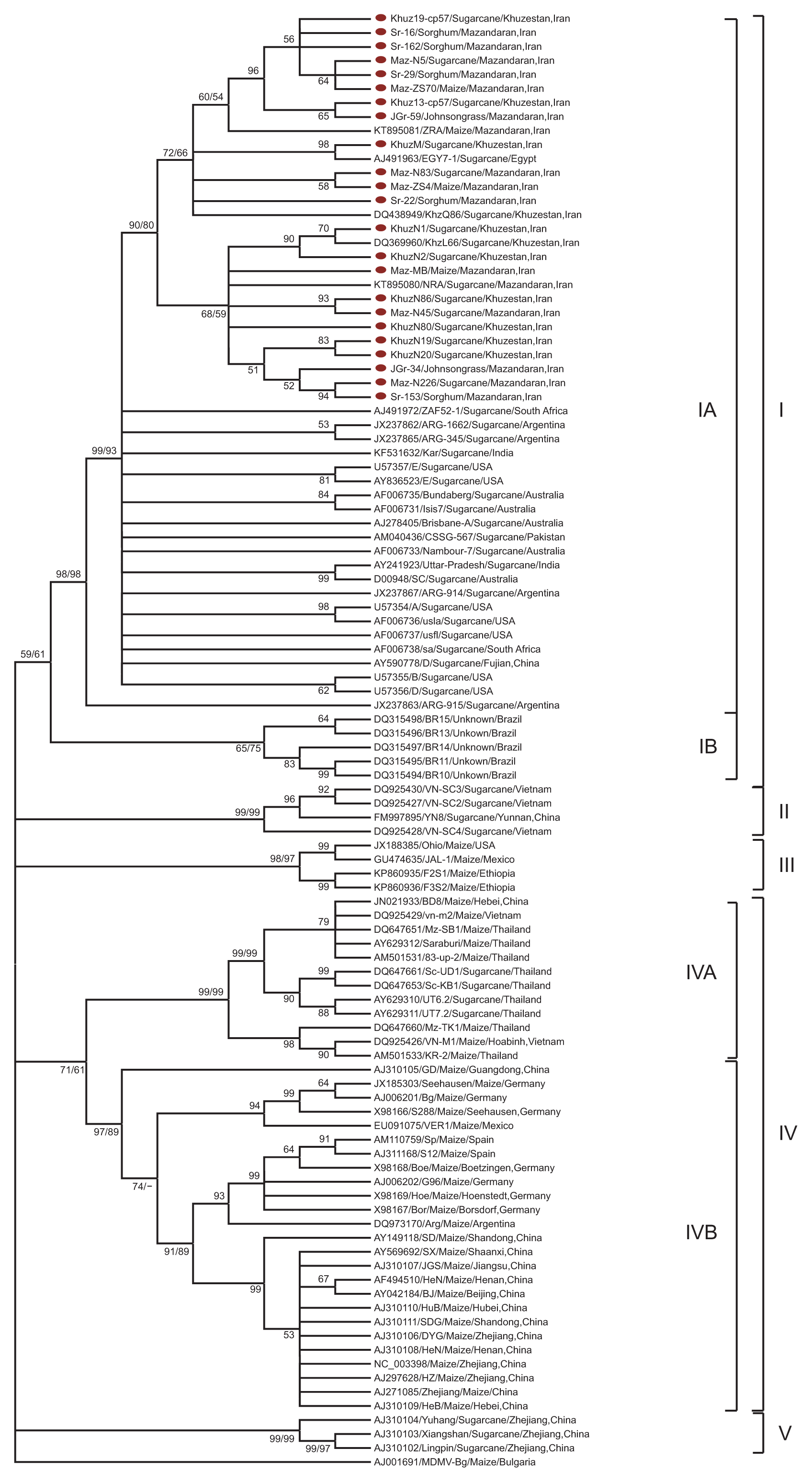
A neighbor-joining (NJ) phylogenetic tree of nucleotide sequences of Sugarcane mosaic virus (SCMV) coat protein genes characterized in this study and their counterparts in the GenBank database. The tree is based on pair-wise alignment, done using ClustalW program of MEGA v.6. Maize dwarf mosaic virus-Bg (AJ001691) was used as an outgroup in the analysis. The percentage of replicate tree in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. Bootstrap values exceeding 50% are given at the major respective nodes obtained from both neighbor-joining and maximum-likelihood methods (NJ/ML %). New sequences are without GenBank codes, in marked, but other sequences are indicated in the tree by accession number/isolate name/host origin/geographical origin of the collection.
Tests of neutrality
DnaSP version 5.10.01 (Librado and Rozas, 2009) was used for testing Tajima’s D (Tajima, 1989), Fu and Li’s D* and F* (Fu and Li, 1993) tests of neutrality. Tajima’s D statistic measures the departure from neutrality for all mutations in a genomic region (Tajima, 1989). Tajima’s D test is based on the differences between the number of segregating sites and the average number of nucleotide differences. Fu and Li’s D* test is based on the differences between the number of singletons (mutations appearing only once among the sequence) and the total number of mutations. Fu and Li’s F* test is based on the differences between the number of singletons and the average number of nucleotide differences between every pair of sequences (Fu and Li, 1993; Tsompana et al., 2005). The G-test (G) statistic of the McDonald and Kreitman test (MKT) (McDonald and Kreitman, 1991) was used to determine whether synonymous and non-synonymous variations support the hypothesis of adaptive protein evolution between the SCMV phylogroups and whether the divergence in the SCMV phylogroups is caused by natural selection. MKT was also performed using DnaSP v. 5.10.01 (Librado and Rozas, 2009).
Tests of population differentiation
Statistical tests of population differentiation including, Kst*, Z, Snn and FST were calculated using DnaSP v. 5.10.01 (Librado and Rozas, 2009). The Z statistic results from ranking distances between all pairs of sequences. The average ranks for those from within two locations are summed, and the sum is weighted. The Z* statistic is a logarithmic variant of the Z statistic. The frequency with which the nearest neighbors of sequences are found in the same locality is measured by the Snn test statistic, whose values may range from 1 (when populations from different localities are genetically distinct) to 1/2 in the case of panmixia. FST is the coefficient of gene differentiation or fixation index, which measures inter-population diversity, and the absolute value of FST ranges from 0 to 1 for undifferentiated to fully differentiated populations (Hudson, 2000). However, practically, the observed FST is much less than 1, even in highly differentiated populations. Normally, an absolute value of FST > 0.33 suggests infrequent gene flow, while absolute value of FST < 0.33 suggests frequent gene flow (Librado and Rozas, 2009; Rozas et al., 2003). Statistical significance for all tests was established using 1,000 permutations test.
Selection pressure analysis
The number of nonsynonymous substitutions per nonsynonymous site (dN), the number of synonymous substitutions per synonymous site (dS), and the dN/dS ratios for the nucleotide sequences of CP genes were estimated for all isolates in each phylogenetic group using DnaSP v.5.10.01 (Librado and Rozas, 2009). The gene is under positive (or diversifying) selection when the dN/dS ratio is > 1, neutral selection when dN/dS ratio = 1, and negative (or purifying) selection when dN/dS ratio < 1. Diversity indices were also calculated using DnaSP v. 5.10.01 (Librado and Rozas, 2009).
Results
SCMV infection
The occurrence of SCMV was surveyed by detection of the virus in several graminaceous leaf samples collected from fields in two major maize-and sugarcane-growing provinces of Iran. The SCMV CP gene, with the expected size of about 900 bp, was amplified by RT-PCR from most of the samples (223/250; infected/total). From a total of 250 samples collected from sugarcane, maize, sorghum and Johnson grass, 106 (92.2%), 84 (87.5%), 22 (88.0%), and 11 samples (78.5%) were infected by SCMV, respectively. All the surveyed fields were found to be infected by the virus (Table 1). SCMV was detectable by RT-PCR in plants with symptoms of mosaic and streaking, and the virus was not detected in extracts of healthy control plants.

Incidence of Sugarcane mosaic virus infection in symptomatic samples collected from different hosts in the surveyed regions of Iran
Sequence determination and data analysis
The CP sequences of 23 SCMV isolates were determined, analyzed and deposited in the GenBank database (accession nos. KX430773–KX430794 and KX938353) (Table 2). The CP gene was found to be 888 nt long, coding for 296 amino acids in all isolates tested here. The DAG box (Asp-Ala-Gly), which is believed to be crucial for aphid transmission (Atreya et al., 1995), and the conserved R-X43-D motif, which is presumably essential for virion assembly, cell-to-cell and long-distance movement (Dolja et al., 1995; Urcuqui-Inchima et al., 2001), were present in all Iranian SCMV isolates. The four motifs VRGS, MVWCIENGCSP, AFDF, and QMKAAA were also found in the CP of these isolates (Dujovny et al., 2000). The motif MVWCIENGCSP was changed to MVWGIEYGCSP in KhuzN1 and to VVWCIENGFSP in Maz-ZS4 (underlined bases are different).
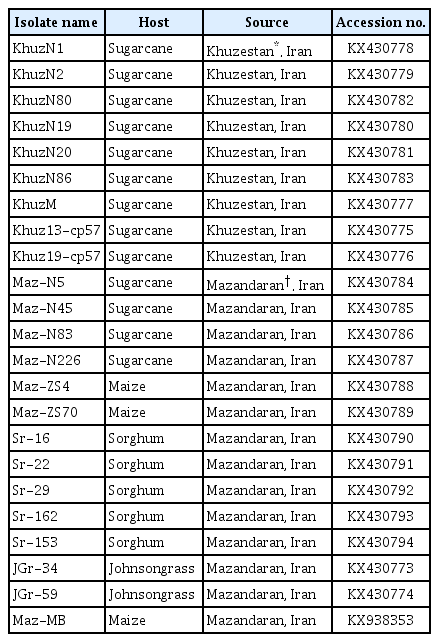
Iranian Sugarcane mosaic virus isolates reported in this study, with their hosts, geographical origins and accession numbers
Sequence comparisons
The pairwise sequence identity of CP genes among all 103 SCMV isolates ranged from 75.2% to 99.9% at the nucleotide sequence level and from 70% to 100% at the amino acid sequence level (data not shown). Comparative sequence analysis of Iranian isolates revealed 96.2–99.9% nt sequence identity among them, indicating a low nt variability. The CP nucleotide sequence identity scores between Iranian and worldwide SCMV isolates ranged between 75.5% (the lowest with Vietnamese isolates VN-SC2 [DQ925427], VN-SC3 [DQ925430]) and 99.9% (the highest with Egyptian isolate EGY7-1 [AJ491963]). Isolates from Egypt and Australia were more closely related to Iranian isolates, with the sequence identity of 97.5–99.9% and 95.8–97.2%, respectively. In the deduced aa sequences, Iranian isolates shared 97.6–100% identity among themselves and 72.8% (the lowest with SC-KB1 from Thailand) to 100% (the highest with EGY7-1) with the SCMV isolates from different countries. Alignment of the CP amino acid sequences of all SCMV isolates revealed that most of the variations in the CP are located in the N-terminus; by contrast, amino acids in the C-terminus were mainly conserved.
Recombination analysis
The CP sequences of 27 SCMV isolates from Iran and those of 76 isolates from GenBank were analyzed for inter- and intra-lineage recombination events. No recombination sites were found in the CP coding region of Iranian SCMV isolates and also other SCMV isolates in subgroup IA. However in some isolates belonging to subgroup IB (including DQ315494/BR10, DQ315495/BR11, DQ315496/BR13, DQ315498/BR15 from Brazil) and group II (including DQ925428/VN-SC4 from Vietnam), recombination was detected. The major and minor parents, beginning and ending breakpoints, event detecting programs and the corresponding P-values are listed in Supplementary Table 1. Among the five recombinants, both Isis7 (AF006731) and BR14 (DQ315497) appeared four times as a parent. All the recombination regions were beyond the highly variable N-terminal region, in accordance with former research (Li et al., 2013).
Phylogenetic analysis and evolutionary divergence
The topologies of both the ML and NJ trees were essentially identical (data not shown). Only the NJ tree with bootstrap values for both NJ and ML is shown in Fig. 1. The results showed that SCMV isolates could be divided into five groups (Fig. 1). Group I included all Iranian isolates (from different hosts) plus 28 sugarcane isolates from Egypt, Australia, Argentina, USA, India, Pakistan, Brazil, South Africa, and China. In this group, the geographical origins of the isolates were the most diverse. Group I could be divided into two subgroups, namely A and B. With the exception of 5 Brazilian isolates (subgroup IB), all other isolates alongside with Iranian isolates fell into subgroup IA. Group II included 4 isolates from sugarcane in Vietnam and China. Group III contained 4 maize isolates from Ethiopia, USA and Mexico. Group IV contained 37 maize or sugarcane isolates from China, Vietnam, Thailand, Spain, Germany and some other countries. Group V included 3 isolates from noble sugarcane in China. The grouping of the 103 SCMV isolates were supported by the subsequent genetic distance analyses (Table 3). The genetic distances of SCMV isolates within and between groups were calculated by the Kimura two-parameter model (Kimura, 1980) in MEGA v.6. The overall mean value of nucleotide sequence diversity was 0.146 ± 0.005. Moreover, the extent of nucleotide sequence divergence within Iranian SCMV isolates was 0.022 ± 0.001. The mean sequence distance of CP genes within group I was 0.052 ± 0.007. The genetic distances within group II was the highest (0.110 ± 0.020), indicating that the isolates in this group were the most genetically divergent, which is in agreement with those of a recent study (Gao et al., 2011). On the contrary, the genetic distance within group V was the lowest (0.031 ± 0.010). However, the mean CP sequence distance between the two groups (II and V) was higher (0.235 ± 0.019) (Table 3). The between-group genetic distances of the five groups were significantly higher than the within-group ones, suggesting that the phylogenetic clustering results of these isolates were reasonable. As shown in the phylogenetic tree, the majority of SCMV isolates are arranged within the five groups irrespective of their country of origin or somehow in their host. Meanwhile, all of the Iranian isolates, all of the Australian isolates and most of the USA and Argentinian isolates were clustered within the group I. This suggests that geographical origin or host plant is not the only determinant factor in the early phylogenetic divergence of SCMV isolates into five main groups. After an Egyptian isolate (EGY7-1), Iranian isolates in subgroup IA displayed the highest nt sequence identity with Australian isolates Brisbane-A, Bundaberg (96.5–97.3%) and SC, Nambour-7, Isis7 (95.8–97.2%). They shared the lowest nt sequence identity (91.4–92.9%) with an Argentinian isolate (ARG-915) in subgroup IA, respectively. In subgroup IA, Iranian isolates shared the highest aa sequence identities of 98.5–100% with isolate EGY7-1 from Egypt, and then with isolates Brisbane-A and Nambour-7 from Australia, ARG-915 from Argentina, CSSG-567 from Pakistan and ZAF52-1 from South Africa with 97.3–98.5% identity.

Genetic distances within and between groups
Population genetic analysis
Analysis of group-specific sequences revealed that members of group IV and II are more diverged than isolates belonging to other groups (Table 4). Negative values of the Tajima’s D, Fu and Li’s D*, and Fu and Li’s F* were obtained for groups I and II, and overall population, indicating an excess of low-frequency polymorphism caused by background selection, genetic hitchhiking, or population expansions (Tsompana et al., 2005). Except for group IV, with negative value of Fu and Li’s D*, values of the Tajima’s D, Fu and Li’s D*, and Fu and Li’s F* for groups III and IV were positive, which arise from an excess of intermediate frequency alleles and can result from population bottlenecks, structure and/or balancing selection (Biswas and Akey, 2006). However, the P-values for Tajima’s D, Fu and Li’s D*, and Fu and Li’s F*-test were not significant (P > 0.10) in all cases (Table 4), indicating that the results were less convincing; also, it is also plausible that purifying selection may be acting on each of the SCMV phylogroups. It was impossible to do these statistical tests for SCMV isolates in group V, as the analysis requires at least four sequences in the DnaSP software (Librado and Rozas, 2009).

Based on the results of MKT, G-test statistics were not significant (P > 0.10) for all groups (with exception of between groups III and V), suggesting that the divergence of sequences belonging to each group from those of another group in the CP gene may be a consequence of random processes. Measurements of genetic differentiation between each SCMV phylogroups provide further support for a significant genetic differentiation between groups based on high values of Wright’s FST (Wright, 1951) (Table 5). Additional support for these results could be observed in significantly high values of Snn, Ks*, Z and Z* statistics (Table 5), indicating that infrequent genetic exchange may have occurred between SCMV populations. Taken together with the results of the phylogenetic and nucleotide diversity analyses, it is reasonable to conclude that global SCMV populations segregated into five groups of sequence variants that are genetically differentiated from one another and show some host and geographical structuring.

Selection pressure analysis
The dN/dS ratio was used to estimate natural selection pressure acting on the SCMV CP gene. The global selection pressure (dN/dS) for all of the sequences was 0.470. This value is less than 1, showing that the SCMV CP gene experiences a negative selection pressure. Also, pairwise comparisons showed that the dN/dS values were significantly less than 1 for most of the CP sequence pairs (P < 0.05; data not shown). The global selection pressure (dN/dS) for all of the Iranian isolates was 0.582. The global dN/dS values for each SCMV group were computed separately and compared. Similarly, a negative selection pressure was estimated for CP sequence pairs within the each group, ranging from 0.358 to 0.587. The CP genes of isolates in the groups I, II, III, IV, and V, had global dN/dS values of 0.587, 0.547, 0.358, 0.472, and 0.498, respectively.
Discussion
At least three viruses, including SCMV, Sorghum mosaic virus, and Sugarcane streak mosaic virus in the family Potyviridae infect sugarcane and cause mosaic disease (Perera et al., 2009; Xu et al., 2008, 2010). As such, the findings of this study suggest that SCMV is a common causative agent of mosaic in sugarcane and maize in Iran. In this study, SCMV was found to naturally infect sorghum and Johnson grass plants grown nearby or some distance away from sugarcane and maize fields. According to RT-PCR results, SCMV was present in 89.2% of the collected samples from all visited areas. The sugarcane varieties under cultivation in Iran are widely infected with SCMV as evidenced by more than 92% infection in our samples (Table 1). This high infection rate could be attributed to vector transmission manner (aphids), vegetative sett planting, and the presence of different host plants (crop and weed). For designing better epidemic control strategies, understanding the important features of RNA viruses, such as the molecular basis of virus geographical range, epidemiological routes, population structure, adaptation to new hosts or emergence of a new virus epidemic is crucial (Elena et al., 2011; Gibbs and Ohshima, 2010; Jones, 2009). For this purpose, the CP sequences of 27 SCMV isolates obtained from different hosts and distant geographic regions of Iran were determined, and their genetic relationships with CP sequences of 76 worldwide isolates of the virus were studied. Pairwise nucleotide sequence identity scores showed that global SCMV CP sequence similarity was between 75.2% and 99.9%. This genetic diversity divided the 103 isolates into five main divergent phylogenetic groups. The rate of genetic diversity and nonsynonymous to synonymous mutations (dN/dS) were significantly different between these five groups. In phylogenetic analysis of CP sequences, Iranian isolates were clustered together with isolates from Egypt, Australia, Argentina, USA, Brazil, India, Pakistan, South Africa, and China. Many studies have been previously done on the phylogenetic relationships of SCMV isolates (Achon et al., 2007; Alegria et al., 2003; Gao et al., 2011; Gemechu et al., 2006; Ha et al., 2008; Li et al., 2013; Wang et al., 2010; Xu et al., 2008). According to previous researches, SCMV isolates clustered in relation to their original hosts, and geographically distinct isolates from sugarcane or maize clustered differently (Li et al., 2013). Interestingly, based on a recent study, the complete genome sequences of two Iranian isolates could not be grouped according to geographical origin or host plant (Moradi et al., 2016) as well as GD (Guangdong, China, AJ310105) (Achon et al., 2007) and Ohio (USA) isolates. In the present study, genetic differentiation and phylogenetic analysis of CP gene of 103 SCMV isolates, consisting of 27 isolates from Iran, revealed that the population structure of the five SCMV phylogroups, were not correlated with their geographical origin or host. With the exception of Iranian isolates, from different hosts, group I consisted of members that originated from sugarcane, while isolates in groups II, III, IV, and V derived from sugarcane, maize, maize/sugarcane, and sugarcane, respectively. Among the five groups in the CP-based phylogenetic tree, groups II, IV, and V were consistent with the reported groups E: SO, C: SCE/MZ, and B: NSCE (Gao et al., 2011), respectively. However, all Iranian isolates and the previously reported isolates in D: SCE group were integrated into group I. Furthermore, isolates in A: MZ group were also integrated into group IV. Such structuring of virus isolates might be related to bottlenecks imposed by the host plant, potential arthropod vectors, and presence of physical and quarantine barriers between countries. Since SCMV can be transmitted through vegetative sett, the germplasm exchange could be another possible explanation for such high sequence similarities observed among the intercontinental isolates of SCMV (Padhi and Ramu, 2011). The higher genetic diversity between groups than within groups of SCMV populations suggests that there is more frequent gene flow within groups than between groups. The study conducted by Li et al. (2013) reported that SCMV populations between maize and sugarcane had relatively frequent gene flow (FST < 0.33); but within maize or sugarcane geography distinct isolates had infrequent gene flow (FST > 0.33). Similarly, no significant difference was detected between maize and sugarcane (also other plants) isolates from Iran (FST < 0.33), indicating that the gene flow between SCMV populations of maize and sugarcane was frequent. However, the close genetic affinities observed between some geographically distant SCMV isolates suggest long-distance migration, probably due to international traffic of propagative sugarcane materials. Historical accounts support the hypothesis that the SCMV isolates were most likely introduced into Iran via importation of sugarcane germplasm materials. For instance, the NRA and ZRA SCMV isolates were introduced into Iran probably from sugarcane-producing countries (Australia, USA, Argentina, India, and Egypt) via contaminated vegetative cuttings (Moradi et al., 2016). Subsequently, further spread of SCMV within Iran could have occurred via movement of contaminated vegetative cuttings across provinces to other host plants such as maize, sorghum, and so on and within provinces via aphid vectors. Mutation and recombination are two main sources of genetic variation in RNA viruses (Gell et al., 2015; Mangrauthia et al., 2008; Moreno et al., 2004; Padhi and Ramu, 2011). Recombination events are common in Potyvirus evolution and can be detected throughout the genome (Chare and Holmes, 2006; Revers et al., 1996; Sztuba-Solińska et al., 2011; Valli et al., 2007). Recombination may play an important role in SCMV evolution, and be an important reason for SCMV genetic diversity (Padhi and Ramu, 2011; Xie et al., 2016). Previously, several recombination events have been reported in SCMV and the probability of recombination was diverse for different genes (Achon et al., 2007; Moradi et al., 2016; Padhi and Ramu, 2011; Xie et al., 2016; Zhong et al., 2005). Based on the CP gene of SCMV, six recombinants were detected (Li et al., 2013). In addition, two isolates from Iran, NRA and ZRA were identified as recombinants in the P1 (with highly significant P-values), HC-Pro, CI, NIa-Vpg, and NIa-pro coding region (Moradi et al., 2016). These results point to the importance of genetic recombination in the evolution of global SCMV populations. However, no signature of recombination was detected in CP gene of Iranian SCMV isolates identified in this study. Natural selection is an important evolutionary mechanism limiting variation of virus populations (Pérez-Losada et al., 2008). In the present study, no significant (P > 0.10) deviation from neutrality was detected for almost any of the groups using MKT. On the other hand, a purifying selection was detected in CP genes of SCMV isolates, enhancing the speed of elimination of deleterious mutations in genes and shaping the stable genetic structure of the population. It is also noteworthy that most of the codons in the CP gene were under negative selection or neutral evolution except for codons 27 and 48, which were under positive selection (Li et al., 2013). In contrast to previous studies, Iranian SCMV isolates were not grouped according to host plant or geographical origin which could be explained by virus transmission characteristics, maize or sugarcane germplasm exchange, continental barriers, host plant, and so on. According to the results, SCMV isolated from maize or sorghum is greatly similar to SCMV isolated from sugarcane. One possibility is introduction of sugarcane from different sources to the country. On the other hand, in Iran, maize and sorghum are frequently planted nearby sugarcane crops and weed grasses such as Sorghum halepense, are commonly found in sugarcane fields. Based on the negative selection, SCMV could constantly accumulate the available variations to adapt to the different hosts. Meanwhile, those plants naturally infected by SCMV could serve as potential reservoirs of the virus in the field. Therefore, avoidance of planting maize near sugarcane crops and control of those weeds are important to mitigate the virus infection. Taken together with this study and previous researches (Alegria et al., 2003; Gao et al., 2011; Li et al., 2013; Xie et al., 2016), differences existed between isolates from different hosts or within the same host from different locations, indicating the complexity of genetic structure of the SCMV populations. Nevertheless, it seems that Iranian SCMV isolates based on complete genomic sequences (Moradi et al., 2016) and CP gene sequences are not grouped according to geographical origin or host plant. Frequent gene flow, negative selection and homologous recombination appear to be important driving forces acting synergistically to limit genetic variation within these SCMV isolates. The present study provides a comprehensive analysis of SCMV population structure based on CP gene and the driving forces involved in its evolution. This information will provide a base for evaluating the epidemiological features of SCMV in Iran and will be suitable in designing long-term, sustainable management strategies for SCMV. Further information of SCMV isolates from other geographic zones and hosts from Iran are needed to fully assess the molecular diversity and evolutionary history of the virus.