Identification of Two Novel Amalgaviruses in the Common Eelgrass (Zostera marina) and in Silico Analysis of the Amalgavirus +1 Programmed Ribosomal Frameshifting Sites
Article information
Abstract
The genome sequences of two novel monopartite RNA viruses were identified in a common eelgrass (Zostera marina) transcriptome dataset. Sequence comparison and phylogenetic analyses revealed that these two novel viruses belong to the genus Amalgavirus in the family Amalgaviridae. They were named Zostera marina amalgavirus 1 (ZmAV1) and Zostera marina amalgavirus 2 (ZmAV2). Genomes of both ZmAV1 and ZmAV2 contain two overlapping open reading frames (ORFs). ORF1 encodes a putative replication factory matrix-like protein, while ORF2 encodes a RNA-dependent RNA polymerase (RdRp) domain. The fusion protein (ORF1+2) of ORF1 and ORF2, which mediates RNA replication, was produced using the +1 programmed ribosomal frameshifting (PRF) mechanism. The +1 PRF motif sequence, UUU_CGN, which is highly conserved among known amalgaviruses, was also found in ZmAV1 and ZmAV2. Multiple sequence alignment of the ORF1+2 fusion proteins from 24 amalgaviruses revealed that +1 PRF occurred only at three different positions within the 13-amino acid-long segment, which was surrounded by highly conserved regions on both sides. This suggested that the +1 PRF may be constrained by the structure of fusion proteins. Genome sequences of ZmAV1 and ZmAV2, which are the first viruses to be identified in common eelgrass, will serve as useful resources for studying evolution and diversity of amalgaviruses.
Common eelgrass (Zostera marina) is a marine monocotyledonous angiosperm that belongs to the family Zosteraceae (Lee et al., 2016). It is one of the most common seagrasses and is predominantly found in temperate coastal waters of the northern and southern hemispheres. Common eelgrasses play important roles in the coastal ecosystem. They form a physical habitat for many marine organisms and act as carbon sinks for long-term carbon storage (Dahl et al., 2016; Reynolds et al., 2016). Seagrasses are believed to have returned to sea at least three times from a terrestrial ancestor (Les et al., 1997). Therefore, they are valuable resources for understanding the mechanisms involved in adapting from a terrestrial to a marine habitat. Several studies using next-generation sequencing for transcriptomic analysis have been performed to investigate the genetic basis of this adaptation, including the role of abiotic stresses such as variable or limited light conditions and salinity tolerance (Kong et al., 2013, 2014; Wissler et al., 2009, 2011).
When total RNA from plant samples was extracted for transcriptomic analysis, viral genome RNAs were isolated together with the host RNAs. As a result, many plant RNA-seq datasets contain associated RNA virus sequences, which can be identified by comprehensive bioinformatic analysis (Kim et al., 2014; Liu et al., 2012; Park and Hahn, 2017a, 2017b). In this study, two novel amalgaviruses were identified in a transcriptome dataset obtained for the common eelgrass (Kong et al., 2014).
Transcriptome dataset of young leaves of common eelgrass (Kong et al., 2014) was downloaded from the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI). The transcriptome data is available under the accession number SRA128272 and contains 11.1 gigabases of paired-end RNA-seq reads. Raw sequence data were screened using the sickle program (version 1.33; https://github.com/najoshi/sickle; parameters, -q 30 -l 50) to collect high-quality reads. De novo sequence assembly was performed using the SPAdes Genome Assembler (version 3.10.0; http://spades.bioinf.spbau.ru) (Bankevich et al., 2012).
To identify the virus-associated contigs in the assembled transcriptome contigs, a local BLASTX search was performed against a custom-built viral RNA-dependent RNA polymerase (RdRp) sequence database, using the following parameters: -evalue 1e-5 -max_target_seqs 1. The representative RdRp database of known RNA viruses was constructed using sequences obtained from the Pfam database (release 30.0; http://pfam.xfam.org). The core viral RdRp motif sequences were collected from 19 Pfam families having the following accession numbers: PF00602, PF00603, PF00604, PF00680, PF00946, PF00972, PF00978, PF00998, PF02123, PF03431, PF04196, PF04197, PF05788, PF05919, PF07925, PF08467, PF08716, PF08717, and PF12426. Finally, a total of 345 non-redundant viral RdRp motif sequences were converted to a BLAST-searchable database.
Two common eelgrass transcriptome contigs were identified that contained RdRp motifs similar to that present in Southern tomato virus (STV) (UniProt accession number, A8R3Y5; Pfam accession number, PF02123). The two contigs showed about 68% identity in their nucleotide sequences, thereby indicating that they were derived from related but distinct viruses. STV is a member of the genus Amalgavirus of the family Amalgaviridae (Sabanadzovic et al., 2009), which suggested that the two contigs were genomes of amalgaviruses or related viruses.
BLAST searches in the NCBI protein database confirmed that both contigs are closely related to plant amalgaviruses, including STV, Blueberry latent virus (BLV), and Rhododendron virus A (RHV-A) (Martin et al., 2011; Sabanadzovic et al., 2009, 2010). Therefore, the two contigs were tentatively named Zostera marina amalgavirus 1 (ZmAV1) and Zostera marina amalgavirus 2 (ZmAV2). The viral genome sequences of ZmAV1 and ZmAV2 were 3383 and 3316 nt in length, respectively, and their annotation information is available in the NCBI nucleotide database under the accession numbers KY783316 (ZmAV1) and KY783317 (ZmAV2) (Table 1).

Summary of amalgaviruses identified in this study
The common eelgrass RNA-seq reads were mapped to the ZmAV1 and ZmAV2 genome contigs using BWA and the variants were identified using SAMtools/BCFtools (Li and Durbin, 2009). There were 13 and 34 polymorphic sites in ZmAV1 and ZmAV2 genome sequences, respectively (Supplementary Table 1, 2). Hence, each of genome contigs is a composite sequence derived from at least two closely related clones.
Amalgaviruses are double-stranded RNA viruses with a single genomic RNA segment and infect various plants (Liu and Chen, 2009; Martin et al., 2011; Sabanadzovic et al., 2009, 2010). Their genomes contain two partially overlapping open reading frames (ORFs), a 5′-proximal ORF (ORF1) that encodes a putative replication factory matrix-like protein and the second ORF (ORF2) that encodes a RdRp motif. ORF1+2, formed by the fusion of ORF1 and ORF2 proteins, is expressed using a +1 programmed ribosomal frameshift (PRF) mechanism (Depierreux et al., 2016; Nibert et al., 2016).
Amalgaviruses share similarities in their genomic organization with the members of family Totiviridae, which infect fungi and protozoa, and are also phylogenetically related to viruses of the family Partitiviridae having several hosts, including plants, fungi, and apicomplexans (Martin et al., 2011). Therefore, it has been proposed that amalgaviruses possibly represent a transitional intermediate between totiviruses and partitiviruses (Krupovic et al., 2015).
Similar to other amalgaviruses, two overlapping ORFs were predicted in the genome sequences of ZmAV1 and ZmAV2. The proximal ORFs (ORF1) of ZmAV1 and ZmAV2 encoded 382 and 395 amino acid (aa) long proteins, respectively. ORF1 proteins showed sequence and structural similarities to ORF1 proteins of other amalgaviruses. ZmAV1 and ZmAV2 ORF1 proteins were predicted to be exclusively comprised of α-helices and random coils, as observed in the other amalgaviruses (Krupovic et al., 2015).
The second protein encoded by amalgaviruses is an ORF1+2 fusion protein that uses a +1 PRF mechanism for proper translation. A +1 PRF motif sequence, UUU_CGN (underline, codon boundary for ORF1; N, any nucleotide; CGN, a rare arginine codon), is prevalent in plant amalgaviruses (Nibert et al., 2016; Park and Hahn, 2017b). The same +1 PRF motif was observed in other RNA viruses such as Zygosaccharomyces bailii virus Z (ZbV-Z) and influenza A viruses (Depierreux et al., 2016; Firth et al., 2012). The ZmAV1 and ZmAV2 genome sequences were predicted to have a putative +1 PRF sequence, UUU_CGU, which matched the consensus motif UUU_CGN (Fig. 1). The predicted ORF2 of ZmAV1 and ZmAV2 started at the nucleotide positions 942 and 943, respectively, which were the first bases after the +1 PRF site.
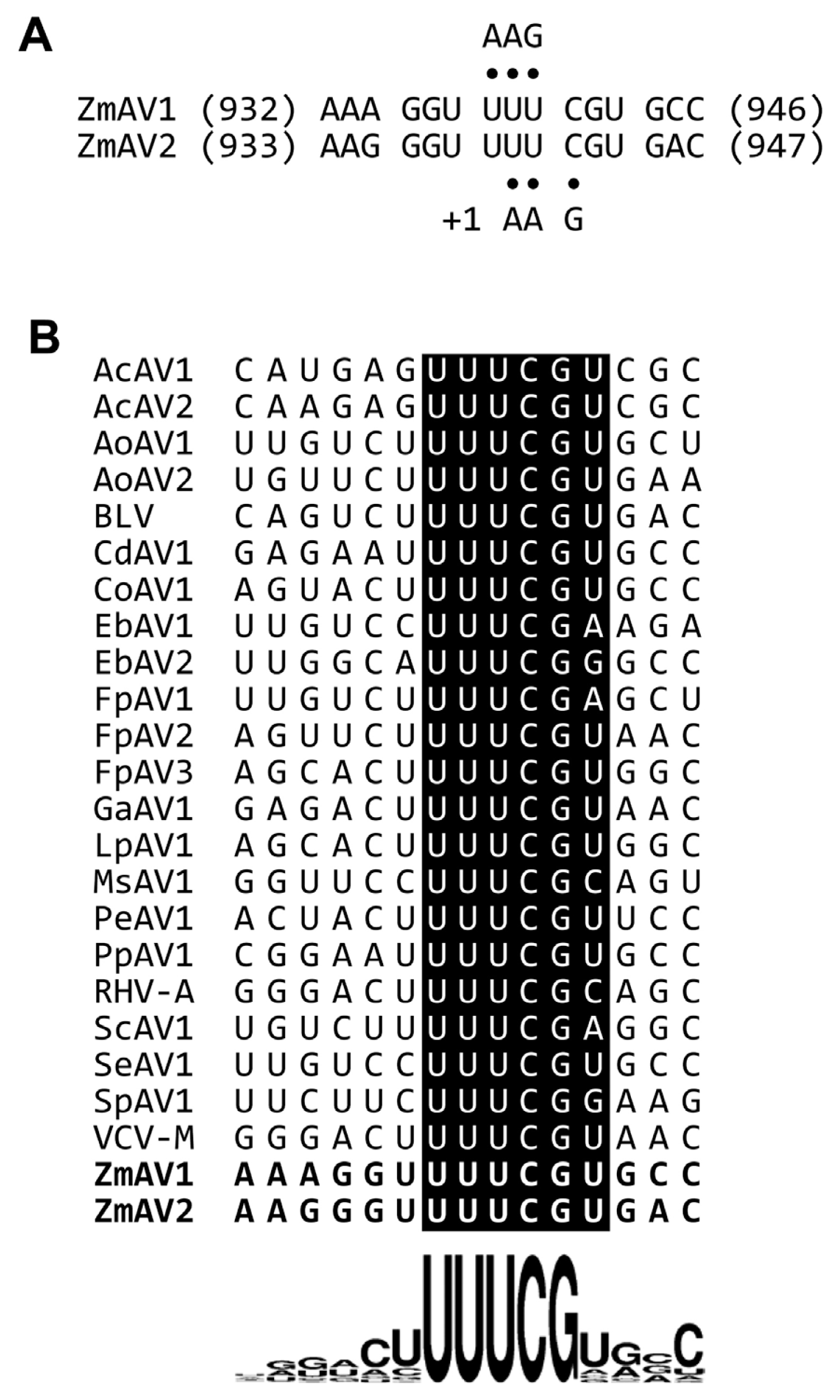
(A) The predicted +1 programmed ribosomal frameshifting (PRF) motifs of ZmAV1 and ZmAV2. Both UUU and UUC codons can interact with the anticodon 3′-AAG-5′ of the phenylalanyl-tRNA (tRNAPhe). The tRNAPhe positioned on UUU is thought to slip forward by one nucleotide, causing a +1 frameshift for continued translation. The codon-anticodon base pairs are marked using dots. (B) Sequence comparison of the +1 PRF motif of amalgaviruses. The ZmAV1 and ZmAV2 +1 PRF motif sequences are marked in boldface letters in the last two rows. Sequence logo representation at the bottom clearly shows the conserved UUU_CGN motif, with uracil (U) being slightly preferred over the other bases before the motif and at the position N of the motif.
The ORF2-encoded peptides in ZmAV1 and ZmAV2 were 785 aa in length and had a conserved viral RdRp motif (Pfam accession number, PF02123). The ZmAV1 and ZmAV2 ORF1+2 fusion proteins produced by the +1 PRF mechanism were 1049 and 1062 aa in length, respectively, and were presumed to mediate viral replication.
The RdRp-motif-containing ORF2 protein sequences of ZmAV1 and ZmAV2 showed 45–50% aa sequence identity with those of the previously reported amalgaviruses (Table 2). The RdRp protein sequence identity threshold for assigning amalgaviruses to different species was 65–70% (Nibert et al., 2016). This indicated that ZmAV1 and ZmAV2 are novel species of amalgaviruses. Furthermore, protein sequence identity between the ZmAV1 and ZmAV2 RdRps was 65.4%, indicating that these two viruses could be considered different species. They showed about 25% aa sequence identity with the RdRp protein of ZbV-Z, the type species of the fungus-infecting genus Zybavirus, which was the most closely related genus to the plant-infecting genus Amalgavirus belonging to the family Amalgaviridae (Depierreux et al., 2016).
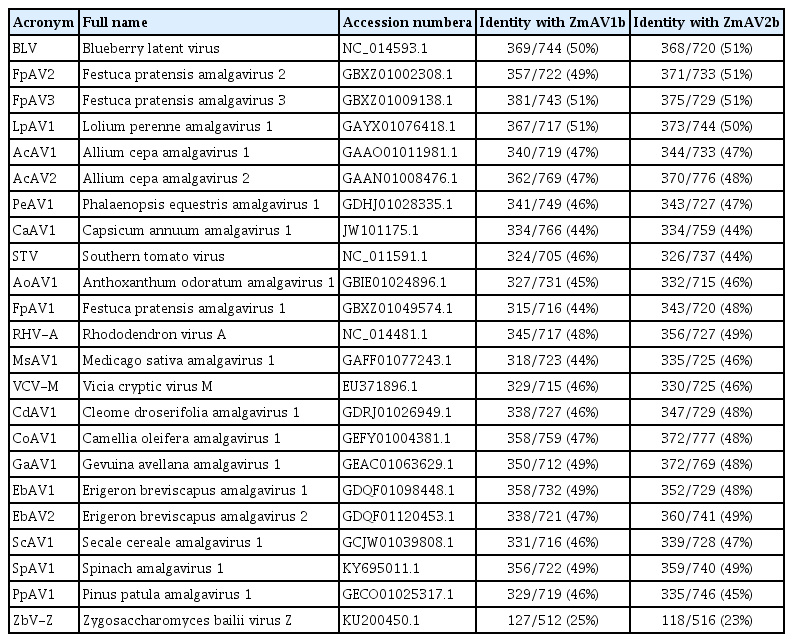
Identities among the RdRp motif-containing ORF2 protein sequences of ZmAV1, ZmAV2, and related viruses
The RdRp-motif containing ORF2 protein sequences of ZmAV1, ZmAV2, and other amalgaviruses were multiply aligned using MUSCLE (https://www.drive5.com/muscle) (Edgar, 2004). Phylogenetic analysis by the neighbor-joining method, using the MEGA7 software (http://www.megasoftware.net) (Kumar et al., 2016) confirmed that ZmAV1 and ZmAV2 are novel closely related species of amalgaviruses (Fig. 2). The observation that ZmAV1 and ZmAV2 formed a distinct clade among known amalgaviruses suggested that a single ancestral amalgavirus infected the common eelgrass in the past and subsequently diverged into two species during the course of evolution. However, it is also possible that two closely related amalgaviruses became independently associated with the common eelgrass.
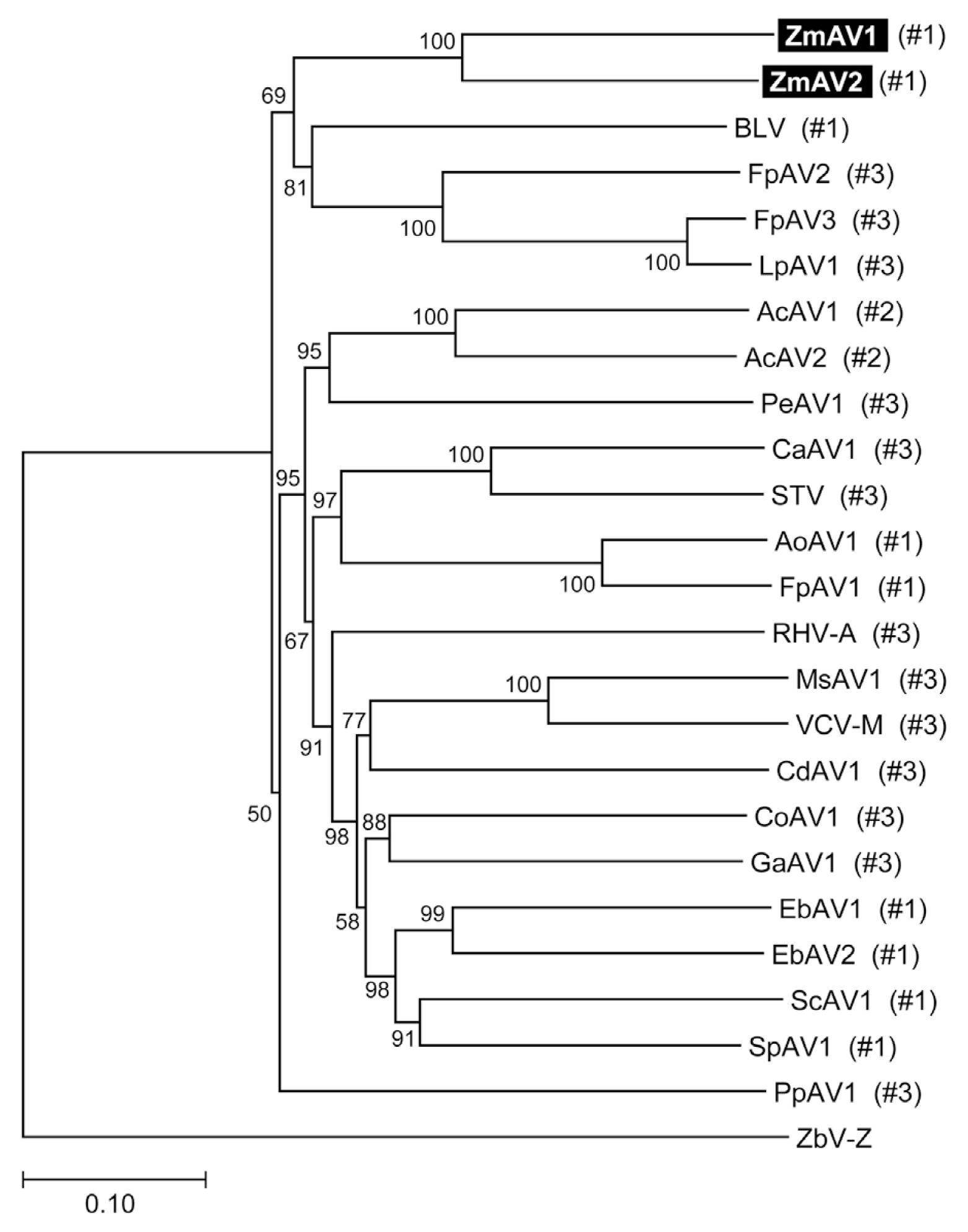
Phylogenetic tree of ZmAV1, ZmAV2, and related plant amalgaviruses. Multiple sequence alignment of the RdRp-motif containing ORF2 protein sequences was performed for inference of the phylogenetic tree. The fungus-infecting virus ZbV-Z was used as an outgroup. The bootstrap values calculated from 100 replicates are shown at the nodes. The +1 PRF site position labels used in Fig. 3A are marked in parentheses.
To analyze whether the +1 PRF site in the fusion protein was conserved, multiple sequence alignment of the ORF1+2 fusion protein sequences of ZmAV1, ZmAV2, and 22 other amalgaviruses was performed (Supplementary Fig. 1). Interestingly, the +1 PRF site of 24 amalgaviruses occurred only at three different positions, which were designated as positions #1, #2, and #3. The +1 PRF occurred at position #1 in 9 amalgaviruses, at position #2 in 2, and at position #3 in 13 (Fig. 3A). Two amalgaviruses, namely STV and Capsicum annuum amalgavirus 1 (CaAV1), had a +1 PRF motif different from those of the other amalgaviruses (Nibert et al., 2016). However, their +1 PRF motif occurred at the same position (position #3) as in the other 11 amalgaviruses.
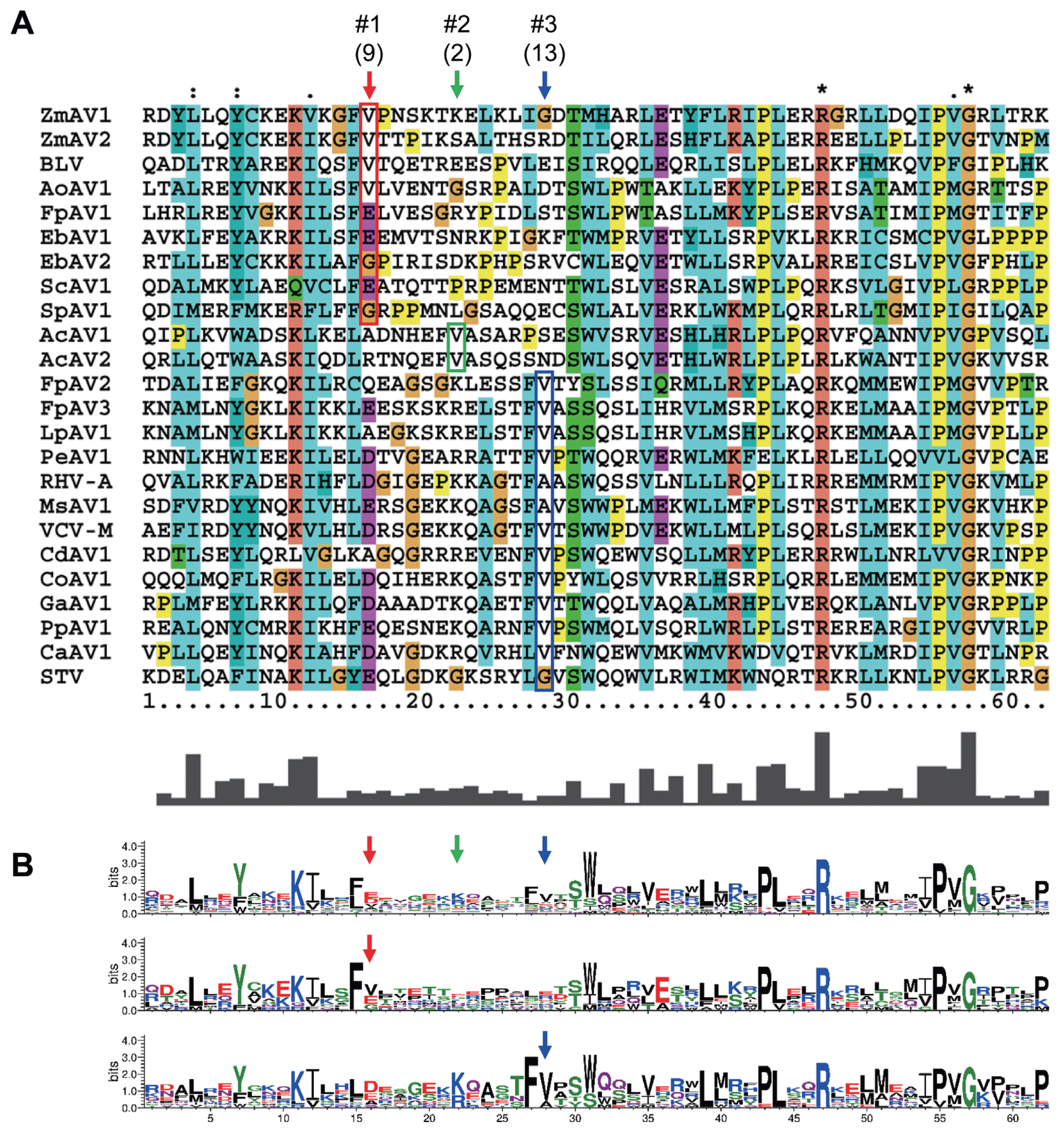
(A) The +1 PRF sites and surrounding regions. Multiple sequence alignment of regions encompassing the +1 PRF sites was performed using the 24 full-length amalgavirus ORF1+2 fusion protein sequences (Supplementary Fig. 1). The +1 PRF occurred at three positions which were labelled as #1, #2, and #3 and marked using red, green, and blue arrows, respectively. Number of viruses are shown in the parenthesis. The first aa residues of ORF2 are shown in colored boxes. (B) Sequence logo representations generated from all viruses (top), and those generated from viruses with the +1 PRF sites at position #1 (middle) and position #3 (bottom). Notably, the +1 PRF sites are embedded in a segment with diverse aa residues and surrounded by conserved regions.
The consensus +1 PRF motif sequence is UUU_CGN, where underline indicates codon boundary for ORF1. Therefore, the last aa residue of ORF1 part of ORF1+2 fusion protein is always a phenylalanine (a UUU codon), except in STV and CaAV1, which have a leucine residue (a CUU codon) at this position because the +1 PRF sequence of these viruses is CUU_AGN (Nibert et al., 2016). When a +1 PRF occurs, the codon boundary changes from UUU_CGN (ORF1) to U_UUC_GNN (ORF2). The first aa residue of ORF2 encoded by a GNN codon can be one of valine, alanine, aspartic acid, glutamic acid, or glycine. All of these aa residues were observed in the 24 fusion proteins (Fig. 3A), suggesting that any aa of them was acceptable at this position. The most frequent base present at the second base position of the GNN codon was uracil (U) (Fig. 1B). Because a GUN codon codes for a valine, the most common residue of the first aa of ORF2 is valine.
The three +1 PRF sites were located within a 13-aa-long segment, which contained diverse residues and was surrounded by many conserved residues (Fig. 3B). Interestingly, the three positions were evenly spaced. There were five residues present between positions #1 and #2 and between #2 and #3. Phylogenetic distribution of the +1 PRF sites revealed that the position shifting event occurred frequently during the evolution of amalgaviruses (Fig. 2). However, only the three specific positions closely located to each other were repeatedly involved. This observation strongly indicated that the +1 PRF site was highly constrained, probably by the folding of fusion proteins. Secondary structure prediction of selected fusion proteins using PSIPRED (version 3.3; http://bioinf.cs.ucl.ac.uk/psipred) (McGuffin et al., 2000) showed that the +1 PRF sites were preferentially located within a random coil segment between two α-helices, one from ORF1 and the other from ORF2, or near the tip of an α-helix. The +1 PRF site seems to be selected not to disrupt the proper folding of ORF1+2 fusion protein.
In conclusion, the full-length genome sequences of two novel amalgaviruses (ZmAV1 and ZmAV2) associated with common eelgrass were identified. Notably, no known viruses have been identified in eelgrasses (genus Zostera) (http://www.genome.jp/virushostdb; as of July 28, 2017) (Mihara et al., 2016). ZmAV1 and ZmAV2 are the first viruses to be identified in eelgrasses. Comparison of the ORF1+2 fusion proteins showed that three +1 PRF sites were preferred, potentially owing to structural constraints in fusion proteins.
Supplementary data
Acknowledgments
This research was supported by the National Research Foundation of Korea funded by the Korea Government (grant number 2017R1A1B400586).