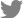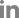Apple scar skin viroid (ASSVd) is one of the smallest viroid pathogens infecting pome fruits, especially apple, and causes a significant damage to fruit tree. It belongs to the genus Apscaviroid, having a circular RNA molecule, 329-330 nucleotides in length (Yang et al., 1992) and a central conserved region within the central domain of the proposed rod-like secondary structure (Flores et al., 1998). ASSVd is generally transmitted through grafting infected scion or pruning tool like secateurs contaminated with crude sap. It is also transmitted through seeds of infected trees at a low rate (Kim et al., 2006). Intense dappling, scarring or cracking is expressed in fruit skins, not in leaves, stems and the other organs, although viroids exist in the vegetative organs (Kim et al., 2006; Koganezawa et al., 2003). ASSVd was first reported at China (Liu et al., 1957), and has been widespread worldwide including Japan (Ushirozawa et al., 1968), USA (Intl. Commit. Coop. Fruit Tree Virus Res., 1985) and Korea (Kwon et al., 2002). Several methods have been developed for detecting viruses and viroids at early stage of infection to fruit trees. The general method for detecting viruses has been indexing, in which the virus is transmitted by grafting or budding to sensitive indicator plants, which then develop identifiable symptoms (Németh, 1986). However, this method requires a lot of time, space, money and experienced labors, and limits the plants to be tested. Enzyme-linked immunosorbent assay (ELISA) using antibodies against specific proteins has widely been used for detecting viruses due to its specificity. Furthermore, ELISA method is not applicable to detect viroids, because they do not possess a coat protein and accordingly no antibodies are available for their detection. However, ELISA often fails because of low virus titers or the inhibitory effects of plant polysaccharides or phenolic compounds (Menzel et al., 2003). Polymerase chain reaction (PCR) has been suggested as an alternative to improve the detection sensitivity and convenience. Especially, multiplex reverse transcription (RT)-PCR has allowed the simultaneous detection of several viruses and viroids in one assay and reduced the costs and time (Hassan et al., 2006). Furthermore, using RT-PCR-probe labeled with digoxigenin, RT-PCR-ELISA system was developed to detect six viroids simultaneously (Shamloul et al., 2002). However, the risk of a carry-over contamination of RNA or DNA still exists in the technique.
Recently, a novel method has been utilized to detect ASSVd, known as reverse transcription loop-mediated isothermal amplification (RT-LAMP) (Lee et al., 2018). This assay amplifies target DNA or RNA with high sensitivity, efficiency and rapidity under the temperature constantly maintained at 62-65°C. There is no requirement for the process of denaturing double stranded DNA prior to amplification and any other measuring equipment and skillful experts. For these reasons, RT-LAMP has potentially been applied for detecting plant viruses and viroids (Lee et al., 2016).
Nucleic acid sequence based amplification (NASBA) method has been introduced for detecting several plant pathogens (Vašková et al., 2004). NASBA is a sensitive transcription-based amplification system for the specific replication of nucleic acids in vitro (Compton, 1991) and based on isothermal amplification which uses two specific oligonucleotide primers and three enzymes of avian myeloblastosis virus reverse transcriptase, RNase H and T7 RNA polymerase, working in concert at a low isothermal temperature, generally at 41°C. This process exponentially amplifies the products producing single-stranded RNA of opposite sense to the original target as the major amplification product (Hibbitts et al., 2003). The amplification products have been detected through a probe-capture hybridization using electrochemiluminescence in molecular beacons.
We have developed real-time quantitative NASBA assay using specific molecular beacon for detecting ASSVd in order to certify disease-free apple cultivars. In this study, the assay and its application for detecting ASSVd in apples (Malus × domestica) are reported and compared to the results from the established RT-PCR.
Materials and Methods
Plant materials
Ten scion cultivars (Beni Shogun, Chukwang, Fuji, Gamhong, Hongro, Hwahong, Jonagold, Sansa, Seokwang and Yoko) and ten rootstock cultivars (M.9, M.9 EMLA, M.9 T337, M.26, M.27, Ottawa 1, Ottawa 2, P.2, P.22 and Pajam 4) grown at the orchard of National Institute of Horticultural and Herbal Science (NIHHS) were used in this study. ASSVd-infected scions were collected from infected mother stock. To avoid uneven viroid distribution, leaves were collected from different parts of randomly selected stems from each cultivar.
Isolation of nucleic acids
Viroid RNA was extracted from infected leaves using the NucliSens® Basic Kit (BioMérieux Inc., Durham, NC, USA) based on the modified method of Boom et al. (1990) as recommended by the manufacturer. The leaves were pulverized in a mortar and pestle using liquid nitrogen and then resuspended with 1 ml of Nucli-Sens® lysis buffer consisting of 5 M guanidine isothiocyanate, 0.1 M Tris-HCl (pH 6.4), 20 mM EDTA, and 1.2% Triton X100 (w/v) with 2% polyvinylpyrrolidone. After centrifugation at 16,000 × g for 15 min, 600 μl of the supernatant was transferred in NucliSens® extractor cartridge supplemented with 50 μl silica suspension. Total nucleic acids were extracted with NucliSens® automatic extractor. Briefly, nucleic acids bound on silica were washed twice with 1 ml NucliSens® wash buffer consisting of 100 mM Tris-HCl (pH 6.4) and 10 M guanidine thiocyanate, twice with 1 ml 70% ethanol and once with 1 ml acetone and dried at 56°C for 10 min. Purified RNA was eluted in 50 μl elution buffer at 56°C for 10 min and stored at −70°C until use.
Primer and molecular beacon design
The ASSVd specific primers, P1 and P2, for amplification of the viroid RNA were designed based on highly conserved regions of selected ASSVd isolates and our sequence of ASSVd isolate collected from ‘Hongro’ cultivar in this study. The alignments were carried out by Clustal W method with Lasergene 7 Megalign (DNAstar, Madison, WI, USA) (Fig. 1). Primers and molecular beacon were designed on the manufacturer’s recommendations using the program Beacon Designer (Premier Biosoft, Palo Alto, CA, USA). Molecular beacon carries a fluorogenic reporter molecule (6-carboxyfluorescein, FAM) at the 5′ terminus, while the arm sequence at the 3′ terminus is covalently linked to a quenching dye (4-(4-4dimethylaminophenylazo)-benzoic acid, DABCYL). Formation of hairpin structure of molecular beacon was predicted by the mfold DNA folding program (Zuker and Stiegler, 1981) for single-stranded DNA. Primers and molecular beacon were synthesized and HPLC-purified. The sequences and positions of the primers and molecular beacon were listed in Fig. 1 and Table 1. A dendrogram was constructed using all sequences of ASSVd isolates from the Genbank of NCBI (accessions AF421195, DQ362906, DQ362907, EU031455, EU031456, U031467, EU031477, EU031487, HG764197, HG764198, G764199, HG764200, HG764201, HG764202, G764203, HG764204, HG764205, HQ840722) with our sequence identified in this study (Fig. 2). The ASSVd sequence which is confirmed in this research belonged to the distinct clade which is contained in ASSVd-K with high similarity. Using MEGA X (Kumar et al., 2018) software, we calculated the genetic distance between ASSVd isolates including our sequence and used the maximum likelihood method based on the Tamura-Nei model with 1,000 bootstraps. In dendrogram, ADFVd (NC_003463), CBLVd (M74065), PBCVd (NC_001830) were used as the outgroup of ASSVd isolates belong to the genus Apscaviroid.
RT-PCR
ASSVd-specific forward and reverse primers reported by Lee et al. (2001) were used for the reverse transcription and PCR amplification of ASSVd RNA molecules. RT-PCR was performed in 50 μl reaction volume containing 5 μl of total RNA, 25 μl of 2 × AccessQuick™ master mix (Promega, Madison, WI, USA), 1 μl of 5 U AMV reverse transcriptase, and 33 pmole each primers. Amplification was carried out in a thermocycler (PTC-220, MJ Research, St. Bruno, Quebec, Canada). Reaction mixture was incubated at 42°C for 70 min. Then PCR was performed at 94°C for 5 min, followed by 40 cycles at 94°C for 45 s, 57°C for 30 s and 72°C for 45 s, and final extension for 5 min at 72°C. Then, the amplicons of target DNA were examined on agarose gel electrophoresis.
Cloning and in vitro transcription
RT-PCR band was excised from the agarose gel and extracted using QIA-quick Gel Extraction Kit (Qiagen, Valencia, CA, USA). PCR product obtained from gel was cloned directly into pGEM-T Easy Vector (Promega, Madison, WI, USA). Plasmids were transformed into competent cells, TOP 10F′ (Invitrogen, Carlsbad, CA, USA) by chemical transformation. Transformants were color-screened on Luria-Bertani medium with 50 μg/ml ampicillin, 0.1 mM isopropyl β-D-1-thiogalactopyranoside and 40 μg/ml X-gal, overnight at 37°C and the presence of the expected inserts confirmed by colony PCR. Plasmids were purified with High Pure Plasmid Isolation Kit (Roche, Basel, Switzerland). These plasmids were verified by sequencing and plasmid containing the inserts was linearized using restriction enzyme ApaI. Transcripts were generated from an upstream promoter using SP6 RNA polymerase, RNasin ribonuclease inhibitor and Riboprobe in vitro Transcription Kit (Promega, Madison, WI, USA) with 1 μg of linearized template DNA in a final reaction volume of 20 μl according to the manufacturer’s instructions. To remove plasmid DNA, RNasefree DNase was treated and the RNA copy number was estimated by UV spectroscopy at an optical density of 260 nm, using 108,968.6 as the estimated molecular weight of each deoxynucleotide and Avogadro’s number to give copies per mole.
Real-time NASBA
Real-time NASBA was carried out in 20 μl reactions using reagents from the NucliSens® basic kit, which is the volume recommended by the basic kit protocol. NASBA reactions were started by mixing 5 μl of extracted nucleic acid and 10 μl of NASBA mix in microtubes. The reaction mixture consisted of 40 mM Tris HCl (pH 8.5), 12 mM MgCl2, 60 mM KCl, 5 mM dithiothreitol, 15% v/v dimethyl sulfoxide, 1 mM dNTPs, 2 mM each of rATP, rUTP and rCTP, 1.5 mM rGTP, 0.5 mM inosine 5′-triphosphate (ITP), 30 μM 3 primer P1, 30 μM primer P2, and 10 μM molecular beacon. To increase the yield of NASBA products, 2 mM ITP was added in the tubes, because of GC-rich and intramolecular base paired structure of ASSVd (Nakahara et al., 1998). Subsequently, the reaction mixtures were incubated at 94°C instead of the standard 65°C for 2 min for denaturation, followed by incubation at 65°C for 5 min and 41°C for 5 min. Then, 5 μl of enzyme mix consisting of 375 mM sorbitol, 2.1 μg bovine serum albumin, 0.08 U RNase H, 32 U T7 RNA polymerase and 6.4 U AMV reverse transcriptase was added to each tube. The tubes were centrifuged briefly in a microcentrifuge to collect the enzyme mix. After tapping gently, the tubes were incubated at 41°C for 150 min. Molecular beacon labeled with FAM was excited at 485 nm, and fluorescence emission was detected at 530 nm.
Results
Primer and molecular beacon for ASSVd
The sequences were aligned for fourteen ASSVd isolates including Korean isolate that we cloned (Fig. 1). Our cloned sequence had 100% homology with the sequence of ASSVd-K (AF421195) reported by Lee et al. (2001). Molecular beacon ASSVd-MB was designed to carry 5′ end and 3′ end sequences of 7 nucleotides complementary to each other and internal sequence of 23 nucleotides complementary to the ASSVd specific NASBA product to hybridize. Secondary structure was confirmed to form hairpin-like loop shape at 41°C using mfold DNA folding program.
NASBA condition
To develop real-time NASBA assay, we designed specific primers and molecular beacon for ASSVd, and then these primers were tested for the best performing reaction. The second step was to find out optimal KCl and ITP concentrations for amplification. For the following steps, twenty samples which were confirmed to be negative for ASSVd were analyzed to define the threshold level for detection. To define the optimal primer concentration, we examined fluorescent signals ranging from 20 to 60 μM (Fig. 3A). The primer concentration with the highest analytical sensitivity or with the highest signal should be regarded as the optimal primer concentration for detecting ASSVd. The optimal primer concentration was found to be 30 μM. Optimal KCl concentration, respectively for corresponding primer pair and ITP concentration was 60 mM (Fig. 3B) and 2 mM (Fig. 3C). This assay was optimized at 60 mM KCl, 30 μM primers, 10 μM molecular beacon and 2 mM ITP. The specific primers target and amplify the sequence from ASSVd found in apple fruits. In order to reflect as reliable as possible whether a given sample is positive or negative for ASSVd, 20 samples confirmed to be negative for ASSVd were analyzed in four different runs using real-time NASBA. From the highest and lowest baseline fluorescence signals, the ratio and average baseline fluorescence could be used to calculate the threshold level. Subsequently, we have decided threshold level for detection of ASSVd at 1.414 (data not shown).
Quantitative real-time NASBA
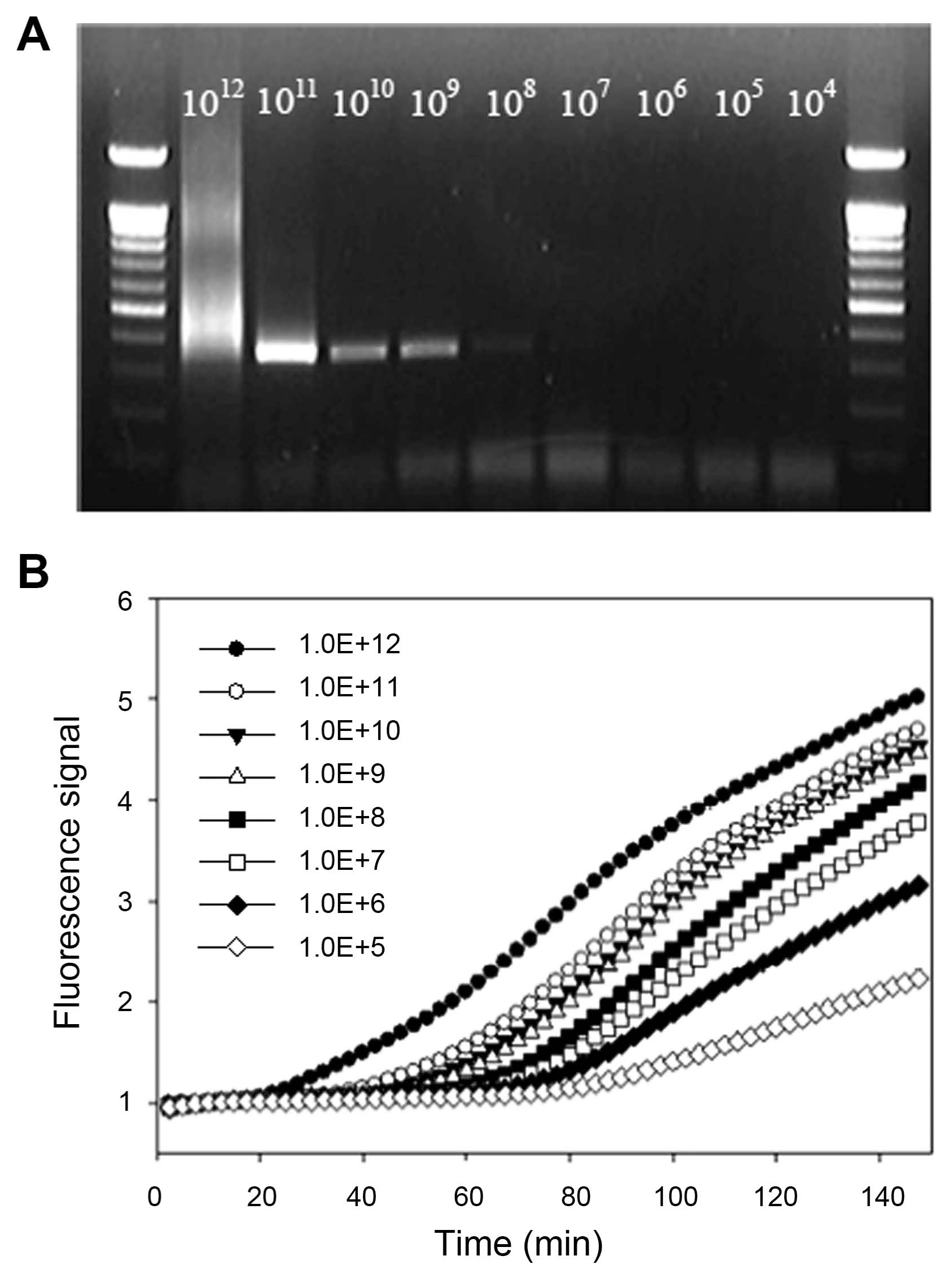
The above developed assay could be applied quantitatively by assuming the amount of unknown RNA samples from the standard curve constructed with the known amount of RNA molecules. To construct standard curve based on time-to-positivity (TTP) values, indicating the cycle number in which amplification first enters a logarithm phase like Ct value in realtime PCR, dilution series of ASSVd RNA standard were prepared ranging from 1×104 to 1×1012 copies/μl. The fluorescence signals of in vitro synthesized ASSVd RNA were shown in Fig. 4. The intervals between TTP values were consistent over a range of at least six orders of magnitude of RNA molecule from 1×1012 to 1×107, as shown by the decimal dilution series. The limit of detection was determined at 1×105 RNA copies for 150 min. A standard regression line was constructed by plotting the TTP values versus the logarithm of the starting ASSVd RNA copy number of 10-fold dilution each. The number of RNA copies present in the reaction was indicated on the X axis and the TTP value in indicated on the Y axis. TTP values were inversely correlated with the initial copy number of ASSVd RNA. The higher the ASSVd RNA, the less reaction time needs to amplify to a detectable level. For the data ranging from 1×107 to 1×1012 copies, the TTP value was the mean of three independent experiments. Starting template copies lower than 1×107 were not consistently amplified. The linear quantification was made between 1×107 to 1×1012 RNA copies (R2 = 0.99, P < 0.0001).
Comparison of real-time NASBA and RT-PCR results
Two different molecular detection systems were compared to verify their performance and analytical sensitivity by using the same diluted RNA samples. General RT-PCR and real-time NASBA were performed according to the established and developed methods above (Fig. 5). With RT-PCR, the detection of ASSVd was obtained up to approximately 1×108 copies. However, the band was smeared at 1×1012 copies/μl and quite obscure in 1×108 fold dilution. On the other hand, relative fluorescence with dilution series by real-time NASBA could be detect up to 1×105 folds and more sensitive than the established RT-PCR method.
Discussion
In this study, we reported NASBA assay with real-time detection of ASSVd by molecular beacon using the NucliSens ® Basic Kit, which is an alternative to the existing detection methods. RNA extraction using NucliSens® Lysis Buffer and Isolation Reagents (silica), which was based on the methods of Boom et al. (1990), was more convenient to isolate RNA than other extraction kits. Especially, this isolation method is easy and speedy during high-throughput processing to even non-experienced experimenters. However, this kit was not manufactured anymore according to the company policy. Therefore, we were utilizing Nucli-Sens® easyMAG® system in the present study.
Real-time NASBA assay developed in this study was optimized to detect ASSVd exquisitely according to the guidance in development of in-house NASBA assay of manufacturer’s recommendations. The assay mixture was composed of 60 mM KCl, 30 μ 3 primers, 10 μM molecular beacon and 2 mM ITP. Additional ITP was known to increase the efficiency to swell NASBA products which have GC-rich and intramolecular base-paired viroid RNA (Nakahara et al., 1998). Substitution to “I” from “G” in the sequences was not a serious problem to real-time detect by using molecular beacon.
Real-time NASBA assay was compared to parallel RT-PCR assay with respect to detection limits. The results showed that the real-time NASBA assay developed was more sensitive and specific than RT-PCR analysis for the detection of ASSVd in apple trees. Furthermore, this method was faster to perform than RT-PCR and more reliable to detect ASSVd. A set of RNA samples could be analyzed within 2.5 h by real-time NASBA, faster than RT-PCR method combined with gel electrophoresis and imaging. The employment of molecular beacon permitted precise diagnosis in a single tube, without electrophoresis on gel, imaging and contamination risk. RT-PCR could detect as little as 1×108 copies/μl, and was able to analyze in 1×1011 copies/μl of in vitro synthesized RNA. However, PCR band was smeared at 1×1012 dilution, because too many RNAs were included in samples. In the case of 1×108 fold dilution, the ASSVd band was barely visible, because RNA was not sufficient to reverse-transcribe or to amplify by polymerase. On the other hand, the real-time NASBA with molecular beacon could detect from 1×1012 to 1×105 copies of ASSVd RNA. The fluorescence signal at 1×104 fold dilution could also be detected within 150 min (data not shown), even with low reproducibility. ASSVd in samples could be quantified by comparing the TTP value of each sample to the TTP values from the standard regression lines. The amount of starting template in any NASBA reaction, expressed as the copy number of the target ASSVd RNA in the original plant extract, could accurately be determined. The limit of quantification (LOQ) for detection of ASSVd was determined at 1×107 RNA copies from the results. According to this result, routine diagnosis through above real-time NASBA had better to perform within 90 min than 150 min to detect ASSVd, resulting from range of LOQ. The real-time NASBA assay is easier to use and more specific and faster to detect ASSVd in apple trees compared to RT-PCR assay. Furthermore, this assay might especially be useful for detecting low levels of viroids in apple trees at early infection stage and for monitoring their influence on tree growth. Therefore, this assay might be suitable for high-throughput diagnosis and automation like quarantine, certification and breeding programs for viroid resistance in apple trees.


 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print