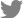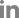With the worldwide increase in trade among nations during last two decades, the number of exotic plants, including crops, fruits, and vegetables, that have been imported into Korea, has dramatically increased. Preventing the entry or spread of exotic pathogens that frequently accompany this plant material into Korea and into other countries relies in part on quarantine inspections. Although the Animal, Plant and Fisheries Quarantine and Inspection Agency (QIA) in Korea attempts to prevent the entry of all quarantined pathogens in imported plants, the identification of exotic plant pathogens and especially of viruses can be difficult because the host range of many plant viruses includes both known and unknown plant species. In addition, plant viruses may be present but may cause disease symptoms only under specific conditions. It is, therefore, important that quarantine agencies must have the ability to detect and identify plant viruses in both symptomatic and symptom-less plants of all plant species.
In this study, we developed an improved method for the detection of five quarantine viruses in Korea: Beet black scorch virus (BBSV), Beet necrotic yellow vein virus (BNYVV), Eggplant mottled dwarf virus (EMDV), Pelargonium zonate spot virus (PZSV), and Rice yellow mottle virus (RYMV). Although these viruses have not been reported in Korea, they could infect economically important plants in this country if introduced. BBSV and BNYVV cause rhizomania disease of sugar beet (Jiang et al., 1999; Tamada and Kusume, 1991). Interestingly, both viruses are transmitted by fungal vectors; BBSV is transmitted in a non-persistent manner by zoospores of Olpidium brassicae (Jiang et al., 1999) while BNYVV is transmitted by the soil-borne fungus Polymyxa betae (Tamada and Kusume, 1991). BBSV is a member of the genus Necrovirus, and its genome consists of 3,641 bp that encode six proteins (Cao et al., 2002). BNYVV is a member of the genus Benyvirus and is composed of four or five RNA segments (Kiguchi et al., 1996; Saito et al., 1996). Although BBSV and BNYVV have not yet been reported in Korea, they have been reported in most other parts of the world (Cai et al., 1993; Chiba et al., 2011; González-Vázquez et al., 2009; Koenig and Valizadeh, 2008; Weiland et al., 2006). Mechanical inoculation demonstrated that BBSV has a wide host range and produces local lesions on Chenopodium amaranticolor, C. quinoa, C. murale, Spinacia oleracea, and Tetragonia expansa (Cai et al., 1993). BBSV also latently infects Lactuca sativa, Physalis florisana, and Nicotiana species (Cai et al., 1993). Aside from sugar beet, BNYVV also infects various spinach cultivars (Mou et al., 2012).
EMDV is a rhabdovirus, transmitted by a leafhopper (Babaie and Izadpanah, 2003) and infects many plants such as Capparis spinosa, cucumber, eggplant, tomato, Pittosprum tobira, potato, and tobacco (Katis et al., 2011; Mavriè et al., 2006). PZSV, belongs to the family Bromoviridae, was originally isolated from Pelargonium zonale and has a wide host range that includes tomato, globe artichoke, Capsella bursapastoris, Chrysanthemum segetum, Diplotaxis erucoides, Picris echioides, and Sonchus oleraceus (Finetti-Sialer and Gallitelli, 2003; Quacquarelli and Gallitelli, 1979). The genome of PZSV consists of three RNA segments, one of which encodes a polypeptide with a molecular mass of 108 kDa (Finetti-Sialer and Gallitelli, 2003). RYMV was first reported in Kenya in 1966 (Hebrard et al., 2009) and so far has been detected only in Africa. The host range of RYMV includes two cultivated rice species, Oryza sativa and O. glaberrima, as well as some wild grasses (Allarangaye et al., 2007). RYMV is a member of the genus Sobemovirus, encodes four proteins, and is transmitted by various beetles belonging to the Chrysomelidae family (Bakker, 1974; Fargette et al., 2002; Yassi et al., 1994).
Since these five viruses have relatively wide host ranges, it is likely that their introduction would result in their establishment and spread among vegetables and weeds in Korea. To accurately and rapidly detect RNA viruses, researchers have developed methods based on reverse transcription-polymerase chain reaction (RT-PCR) (Lee et al., 2004; Lee et al., 2011a, 2011b). In the current study, we designed RT-PCR primer sets for the detection of the five quarantine viruses described in the previous paragraphs. We also demonstrate the utility and specificity of these primer sets.
Background information for the five viruses is provided in Table 1. Virus-infected plant tissues were obtained from DSMZ GmbH in Germany (http://www.dsmz.de/) with strict control by the QIA. To design RT-PCR primer sets specific for each virus, we used the genes encoding the capsid protein (CP) because these nucleotide sequences are conserved within viruses. Nucleotide sequences for each virus were downloaded from the National Center for Biotechnology Information (NCBI). Three primer sets (designated #1, #2, and #3) were designed for each of the five viruses (Table 2).
Total RNA was extracted using Isol-RNA lysis reagent (5 Prime, Gaithersburg, USA) following the manufacturer’s instruction. To determine the quality and quantity of the isolated total RNAs, we loaded the extracted RNA samples on 1% TAE agarose gel; after electrophoresis, the RNA was visualized by ethidium bromide (EtBr) staining, and the RNA concentration in each sample was measured with the Nanophotomer™ (Implen GmbH, Munich, Germany). The extracted total RNAs were of high quality and of sufficient quantity to synthesize cDNAs (Fig. 1). The first strand cDNA was synthesized as described previously (Lee et al., 2011a, 2011b). Briefly, 1 μg of each total RNA, 10 pmoles of each reverse primer, and sufficient DEPC-treated water were combined to a total volume of 5 μl. The samples were kept at 70.0°C for 5 min and then in ice for 5 min. After 5 μl of 2.5 mM dNTP, 5 μl of 5× GoScript™ reaction buffer, 1.5 μl of 25 mM MgCl2, 1 μl of GoScript ™ reverse transcriptase (Promega, Madison, USA), and 3.5 μl of DEPC-treated water were added, the samples were incubated at 42°C for 1 h. The reverse transcription reaction was stopped by incubating samples at 70°C for 15 min.
PCR was conducted in 50 μl of PCR mixture containing 1 μl of five-fold diluted cDNA, 2 μl of each primer set at 10 pmole 5 μl of 10× PCR buffer, 1 μl of 2.5 mM dNTP, 0.25 μl of Ex-Taq polymerase (Takara, Otsu, Japan), and 40.75 μl of dH2O; in the first PCRs, only primer set #1 for each virus (see Table 2) was used. The PCR reactions were carried out with a C1000™ thermal cycler (BIO-RAD, Hercules, USA) as follows: an initial denaturation at 94°C for 5 min; followed by 30 cycles of denaturation at 94°C for 30 sec, annealing at 50°C for 30 sec, and extension at 72°C for 30 sec; and a final extension at 72°C for 5 min. A 5-μl quantity of amplified PCR product was examined in a 1% TAE agarose gel, which was stained with EtBr for UV visualization. PCR products of the expected sizes were obtained for the five primer sets (Fig. 2; lanes 1, 4, 7, 10, and 13), and the nucleotide sequences of each amplified PCR fragment were confirmed by DNA sequencing.
When we conducted RT-PCR reactions with all 15 primer sets and the identical conditions described in the previous paragraph, all primer sets amplified PCR products of the expected sizes except for primer set RYMV_#3 (Fig. 2; lane 15). However, the quantities of amplified PCR products were less with EMDV_#2, EMDV_#3, and PZSV_#3 than with the other primer sets, indicating that annealing temperature or other conditions for RT-PCR amplification might not be optimal for these three primer sets.
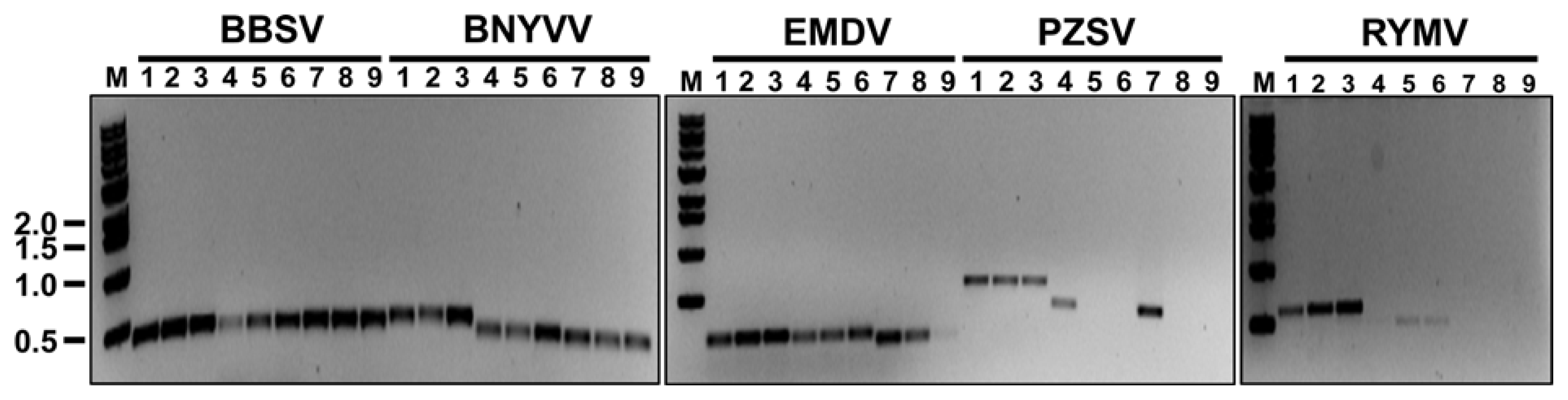
To optimize annealing temperature for each primer set, we performed gradient PCRs with all 15 primer sets. The gradient PCRs were conducted with the following conditions: an initial denaturation at 94°C for 5 min; followed by 25 cycles at 94°C for 30 sec, a gradient annealing temperature (51.9, 54, or 55°C) for 30 sec, and 72°C for 30 sec; and a final extension at 72°C for 5 min. The results are shown in Fig. 3. With primer sets BBSV_#1 and BBSV_#3, clear and substantial bands were obtained regardless of annealing temperature. With primer set BBSV_#2, however, the quantity of PCR product gradually increased as the annealing temperature increased. For BBYVV, PCR products were obtained with all primers at each annealing temperature tested but PCR bands were thicker at 55°C than at other temperatures. Expected bands were obtained with EMDV primer sets at all three annealing temperatures but the band was faint with primer set EMDV_#3 at 55°C. Among the three primer sets for PZSV, only PZSV_#1 amplified PCR products at all three temperatures. In contrast, primer sets PZSV_#2 and PZSV_#3 amplified PCR products only at 51.9°C. For RYMV, only primer set RYMV_#1 could amplify PCR products at all three tested temperatures. With primer set RYMV_#2, amplicons with faint bands were detected only at 54°C and 55°C. The primer set RYMV_#3 did not amplify any PCR product at the three temperatures, indicating that this primer set did not bind to the RYMV cDNA.
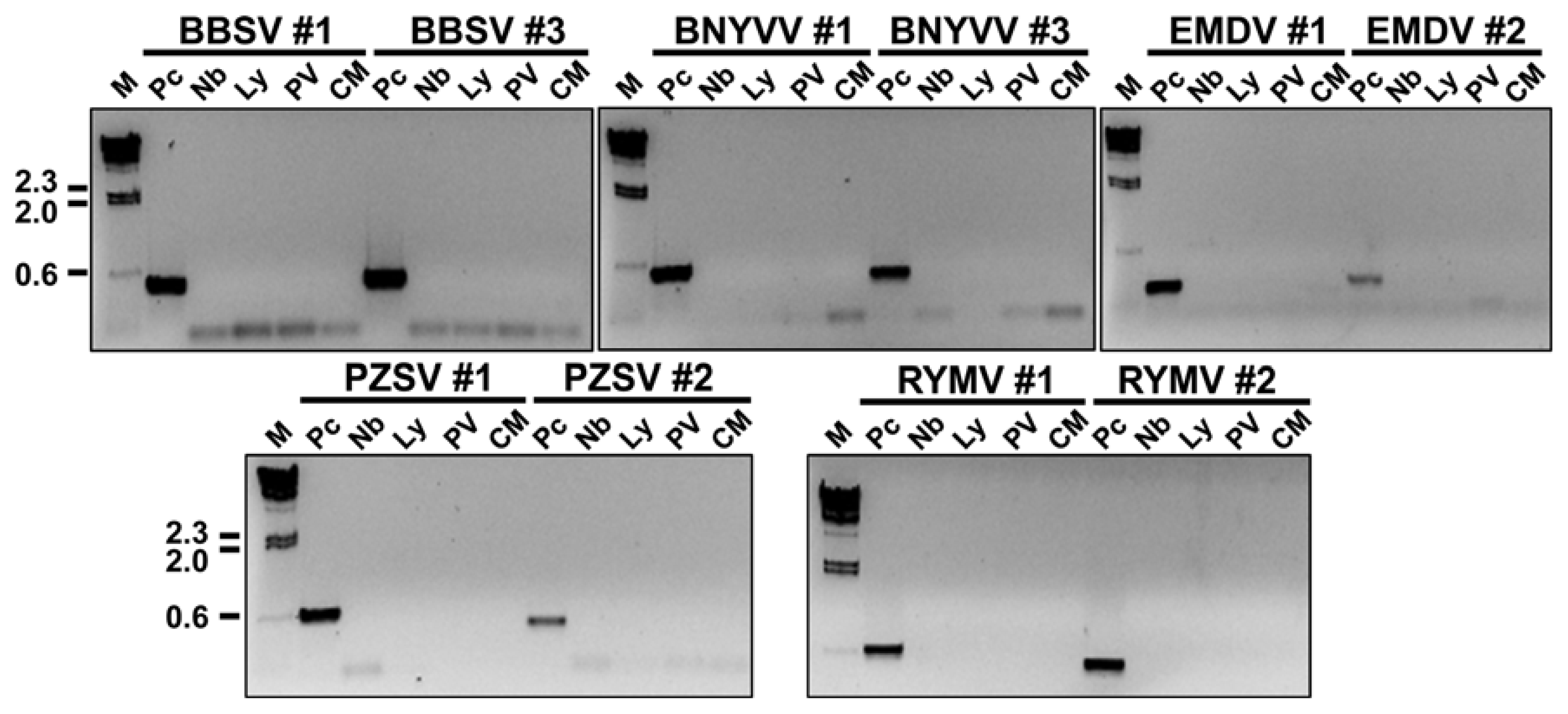
To be useful for quarantine purposes, the designed primer sets must not amplify sequences from other viruses or from the host genome. The specificity of 10 of the primer sets (BBSV_#1, BBSV_#3, BNYVV_#1, BNYVV_#3, EMDV_#1, EMDV_#2, PZSV_#1, PZSV_#2, RYMV_#1, and RYMV_#2) was tested using total RNA of tomato and tobacco and RNA obtained from Nicotiana benthamiana plants that had been inoculated with Potato virus X (PVX) or Cucumber mosaic virus (CMV) 7 days earlier. Total RNAs were extracted from the uninfected tomato and tobacco plants as well as the infected N. benthamiana by the Trizol method, and cDNAs were synthesized as described earlier. PCRs were performed with cDNAs of the five quarantine viruses that were the focus of this study (as positive controls), with the genomic DNAs of N. benthamina and Solanum lycopersicum, and with the cDNAs derived from PVX- and CMV-infected N. benthamiana. PCRs with the 10 primer sets were performed as follows: an initial denaturation at 94.0°C for 5 min; followed by 25 cycles at 94.0°C for 30 sec, 50.0°C for 30 sec, and 72.0°C for 30 sec; and a final extension at 72.0°C for 5 min. PCR products were obtained only with the positive controls (Fig. 4). No band was detected in samples containing genomic DNA of tobacco or tomato, and no band was detected with the cDNAs derived from PVX- or CMV-infected plants. These results indicate that the designed RT-PCR primer sets are specific for the target viruses.
In summary, we designed 15 primer pairs for PCR detection of five viruses (BBSV, BNYVV, EMDV, PZSV and RYMV). These five viruses, which have relatively wide host ranges, have not been detected in Korea but would cause substantial economic losses if they were able to establish in Korea. Fourteen of the 15 primer pairs (all except RYMV_#3) amplified the expected PCR products. Gradient PCRs indicated that 51.9°C was the optimal temperature for the 14 primer sets. Primer pairs BBSV_#1 and BBSV_#3 performed well regardless of annealing temperature while primer pairs PZSV_#2 and PZSV_#3 required a specific annealing temperature. A primer specificity test using plant genomic DNAs and cDNAs derived from the other plant viruses indicated that the primer sets were specific for the five target viruses. Taken together, the results indicate that the primer sets described here will be useful for detecting BBSV, BNYVV, EMDV, PZSV, and RYMV and preventing their entry into Korea.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print