Specific and Sensitive Detection of Venturia nashicola, the Scab Fungus of Asian Pears, by Nested PCR
Article information
Abstract
The fungus Venturia nashicola is the causal agent of scab on Asian pears. For the rapid and reliable identification as well as sensitive detection of V. nashicola, a PCR-based technique was developed. DNA fingerprints of three closely related species, V. nashicola, V. pirina, and V. inaequalis, were obtained by random amplified polymorphic DNA (RAPD) analysis. Two RAPD markers specific to V. nashicola were identified by PCR, after which two pairs of sequence characterized amplified region (SCAR) primers were designed from the nucleotide sequences of the markers. The SCAR primer pairs, designated as D12F/D12R and E11F/E11R, amplified 535-bp and 525-bp DNA fragments, respectively, only from genomic DNA of V. nashicola. The specificity of the primer sets was tested on strains representing three species of Venturia and 20 fungal plant pathogens. The nested PCR primer pair specific to V. nashicola was developed based on the sequence of the species-specific 525-bp DNA fragment amplified by primer set E11F/E11R. The internal primer pair Na11F/Na11R amplified a 235-bp fragment from V. nashicola, but not from any other fungal species tested. The nested PCR assay was sensitive enough to detect the specific fragment in 50 fg of V. nashicola DNA.
Pear scab is one of the most serious diseases affecting both Asian (Japanese pear, Pyrus pylifolia var. culta; Chinese pear, P. ussuriensis) and European (P. communis) pears worldwide. Venturia nashicola is the causal agent of Asian pear scab (Tanaka and Yamamoto, 1964), whereas it is not pathogenic to European pear. Meanwhile, the causal fungus of European pear scab is V. pirina (Shabi, 1990). Because these two fungal species are genetically similar with minute phenotypic differences, some researchers considered that V. nashicola might be a synonym of V. pirina (Shabi et al., 1973). To determine whether or not V. nashicola could be considered a distinct species, morphological examination, mating experiments, and pathogenicity tests were performed and the results revealed that V. nashicola is a species distinct from V. pirina (Ishii and Yanase, 2000). Another genetically related Venturia spp., V. inaequalis, was shown to be the causal agent of apple scab. Sequence analysis of the internal transcribed space (ITS) regions of rDNA revealed that the phylogenetic tree position of V. inaequalis is near the monophyletic group consisting of V. nashicola and V. pirina (Schnabel et al., 1999).
Detection and monitoring of scab pathogen are necessary for the implementation of efficient control strategies. In addition, as Asian pear scab is a quarantined disease in many countries, methods for the rapid and sensitive detection of V. nashicola are required. Recently, molecular methods based on the analysis of DNA sequences, such as ITS regions of the nuclear ribosomal RNA gene, have been widely applied to the identification and detection of plant pathogenic fungi. A species-specific PCR primers designed within the ITS1 and ITS2 regions have been developed to detect plant pathogenic fungi (Asano et al., 2010; Chen et al., 2006; Grote et al., 2002; Ma and Michailides, 2002; Ni et al., 2012). Further, PCR-based random amplified polymorphic DNA (RAPD) is a frequently applied molecular approach for the identification of inter- and intra-species specific molecular markers (Williams et al., 1990). However, as RAPD markers are difficult to reproduce, they should be converted into sequence characterized amplification region (SCAR) markers. Many SCAR primers were designed from the nucleotide sequences of the RAPD markers and have been used for the identification of economically important fungal pathogens (Chioccetti et al., 2001; Grosch et al., 2006; López-Mondéjar et al., 2012; Rigotti et al., 2002; Li et al., 2008; Vandemark et al., 2000).
The aim of this work was to develop species-specific SCAR primers that can detect and differentiate V. nashicola from genetically similar fungal species, especially from V. pirina and V. inaequalis, based on the RAPD markers.
Materials and Methods
Fungal strains
Fungal strains used in this study are listed in Table 1. Among them, nine isolates of V. nashicola were collected from different regions in Korea.

List of fungal strains used in this study
Genomic DNA extraction
Fungal strains were grown on potato dextrose agar (PDA) medium at 25 °C. Genomic DNA was extracted from freshly harvested fungal mycelium using a DNeasy Plant Mini Kit (Qiagen, USA). Mycelium was ground in liquid nitrogen using a mortar and pestle to make a fine powder and total DNA was extracted following the protocol of the manufacturer. The concentration of DNA sample was determined using a DyNA Quant 200 fluorometer (Amersham Bioscience, USA).
RAPD analysis
RAPD amplification was carried out using 120 arbitrary 10-mer primers purchased from Operon (Operon Biotechnologies, USA) and used as single primer. DNA samples from three closely related Venturia species were amplified with the PCR primers. The reaction mixture consisted of 1 × PCR buffer (10 mM Tris-HCl (pH 9.0) 40 mM KCl, 1.5 mM MgCl2), 0.2 mM of each dNTP, 10 pmol of RAPD primer, 2.0 U of Taq DNA polymerase (Bioneer, Korea), and 10 ng of template DNA in a volume of 25 μl. The amplification was carried out in a DNA Thermal Cycler (Takara Shozo, Japan). The cycle parameters were an initial denaturation at 94°C for 5 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 37°C for 1 min, extension at 72°C for 1 min and final extension at 72°C for 5 min. PCR amplification products were separated on a 2.0% agarose gel in TAE buffer.
Cloning and sequencing of RAPD markers
After RAPD bands unique to V. nashicola were excised from agarose gels, the DNA was purified from gel using an AccuPrep Gel Purification Kit (Bioneer, Korea). The purified DNA fragments were ligated into pGEM-T Easy vector (Promega, USA), after which the recombinant plasmids were transformed into competent Escherichia coli DH5α cells. Selection of transformed clone was carried out based on blue/white selection and digestion with EcoRI restriction enzyme. Plasmid DNA was isolated using a Dyne Plasmid Miniprep Kit (Dyne Bio, Korea). The inserted DNA was sequenced by SolGent Co. (Korea).
Design of SCAR primers and specificity test
Based on the nucleotide sequences of specific fragments of V. nashicola, two pairs of oligonucleotide primers were designed using Primer 3 software (Rozen and Skaletsky, 2000). The designed forward primer D12F and reverse primer D12R were expected to amplify a 535-bp DNA fragment with genomic DNA of V. nashicola. Another primer pair, E11F and E11R, was expected to produce a 525-bp DNA fragment specific for V. nashicola. PCR conditions for amplifying the specific fragments with the newly synthesized SCAR primers were as follows: initial denaturation at 94°C for 5 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing for 30 s, extension at 72°C for 45 s, and final extension at 72°C for 5 min. Annealing temperatures of D12F/D12R and E11F/E11R primers were 55 and 62°C, respectively. Specificity of two primer pairs for V. nashicola were evaluated with nine strains of three Venturia species, nine strains of V. nashicola isolated in different regions of Korea and 20 other plant pathogenic fungi listed in Table 1.
Southern blot analysis
To confirm the specificity of SCAR primers for V. nashicola, DNA hybridization was performed using the amplified fragments as probes. PCR products amplified with SCAR primers were excised from agarose gel and then labeled with digoxigenin. Alkaline transfer and Southern hybridizations were carried out as described by Sambrook and Russell (2001). Probe labeling, hybridization and detection were performed with a DIG High Prime DNA Labeling and Detection Starter Kit II (Roche Diagnostics, Germany) as recommended by the manufacturer.
Nested PCR primers for V. nashicola
To increase detection sensitivity, nested PCR primers were designed from the first round of PCR products amplified using the E11F/E11R primer pair. Under low stringent PCR conditions, conducted with annealing temperature of 52°C, the E11F/E11R primer pair produced a 535-bp fragment with DNA of V. pirina, which is the closest fungus to V. nashicola. The PCR product was cloned and the nucleotide sequence was determined as described above. The nucleotide sequence of the amplified product was aligned with that of V. nashicola. Sequence patterns unique to V. nashicola were used to design the specific internal primer pair. The internal primer pair, Na11F and Na11R, for the second PCR was expected to amplify a 235-bp DNA fragment with V. nashicola. For nested PCR, fungal DNA was first subject to amplification by PCR using the external primers E11F and E11R. Subsequently, the amplified product was used as the template for the second PCR using primers Na11F and Na11R.
Sensitivity of nested PCR assay
To determine the detection limit of nested PCR, genomic DNA of V. nashicola was serially diluted with distilled water to yield a final concentration ranging from 50 pg/μl to 500 ag/μl. Template DNA (1 μl) was first subjected to amplification by a DNA Thermal Cycler (Takara Shozo, Japan) using the external primer pair E11F/E11R. Then, one μl of product from the first PCR was used as template for the second PCR using the internal primer pair Na11F/Na11R. PCR amplification was performed using the parameters described above, except that the annealing temperature was 65°C. Ten μl of PCR product was separated on a 1.5% agarose gel in TAE buffer. Gel was stained with ethidium bromide and photographed under ultraviolet light.
Results
Identification of RAPD markers
Five V. nashicola strains, one strain of V. pirina, and three strains of V. inaequalis were used for comparison and identification of specific bands representing V. nashicola using RAPD analysis. Among 120 primers tested in this study, two primers, OPD-12 (5′-CACCGTATCC-3′) and OPE-11 (5′-GAGTCTCAGG-3′), produced polymorphic profiles that distinguished V. nashicola from V. pirina and V. inaequalis. Species-specific bands of approximately 580 bp and 820 bp in length, respectively, were amplified only from DNA of V. nashicola (Fig. 1).

RAPD fingerprints resulting from PCR amplification of genomic DNA of V. nashicola (lanes 1–5), V. pirina (lane 6), and V. inaequalis (lanes 7–9) using the primers (A) OPD-12 and (B) OPE-11. Fragments indicated by arrows were subsequently cloned and sequenced. M, 100 bp DNA size marker (Bioneer).
Design of the SCAR primers
Two RAPD markers for V. nashicola amplified using OPD-12 and OPE-11 primers were excised from agarose gel and DNA fragments were cloned and sequenced. Comparison of the nucleotide sequences of two RAPD fragments did not match any known sequences available in the GenBank sequence database. Based on the nucleotide sequences of the RAPD markers, two SCAR primer pairs, D12F/D12R (5′-ACAAACAGCCTCCCTCATCT-3′/5′-TAACAAACCCACATAAAGAT-3′) and E11F/E11R (5′-AACATACCTGCCACCCAAAA-3′/5′-AAGGCAAGCGAATCCTCATT-3′), were designed. These primer pairs specifically amplified DNA bands of 535 bp (Fig. 2A) and 525 bp (Fig. 2B) in length, respectively, with V. nashicola DNA. Southern blot hybridization with probe derived from the PCR product using SCAR primers and V. nashicola DNA confirmed the specificity of the primer pairs (Supplementary Fig. S1).
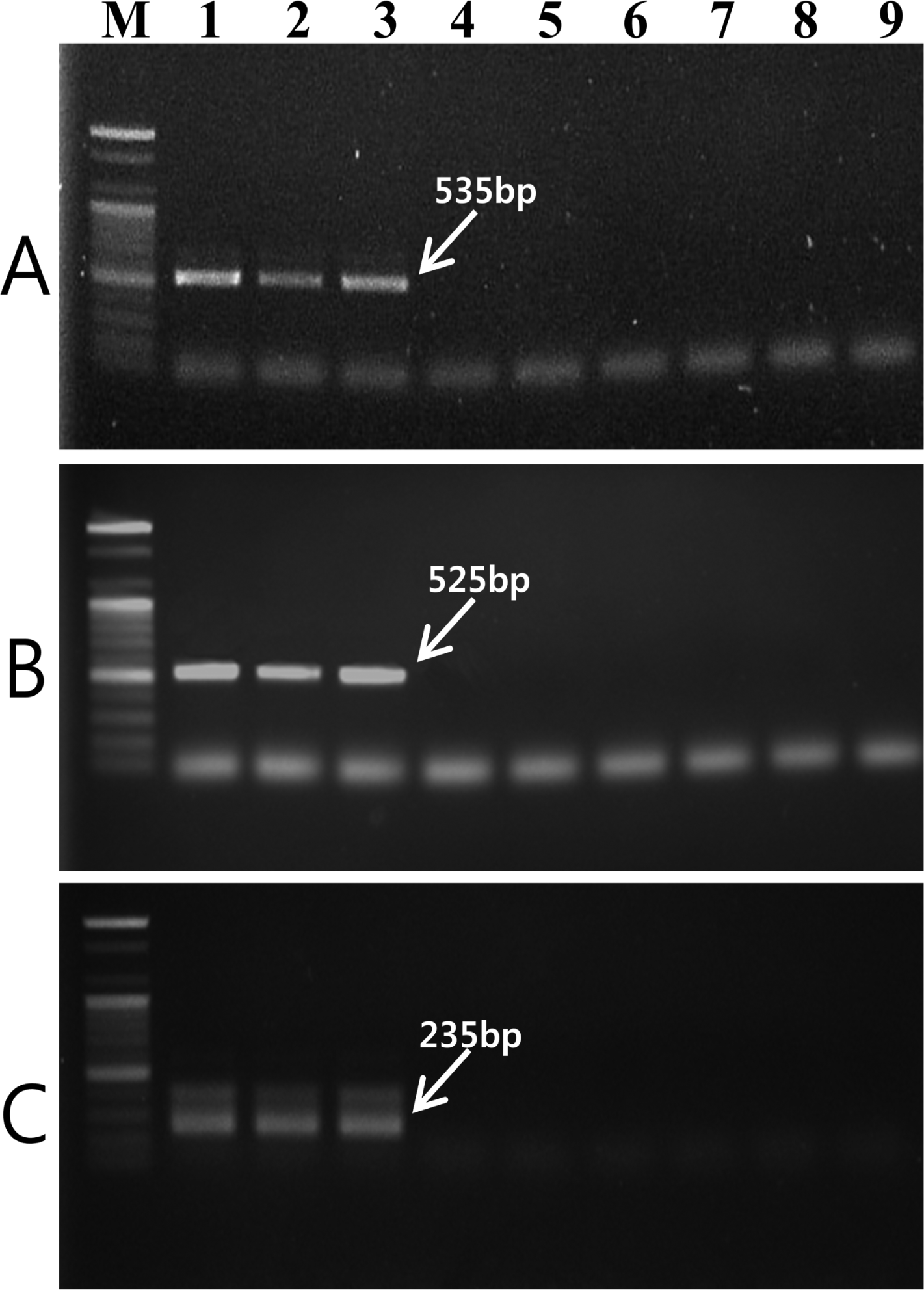
Agarose gel of PCR product amplified with genomic DNA of V. nashicola (lanes 1–3), V. pirina (lane 4–6), and V. inaequalis (lanes 7–9) using (A) D12F/D12R, (B) E11F/E11R, and (C) Na11F/Na11R primer pairs. Lanes 1–3, 4–6, and 7–9 correspond to the strain numbers 1–3, 6–8, and 9–11, respectively, in Table 1. M, 100 bp DNA size marker (Bioneer).
Specificity of primer pairs
To test the specificity of the designed primer pairs, purified DNA samples from 20 plant pathogenic fungal species and nine V. nashicola isolates collected in Korea were amplified using both the D12F/D12R and E11F/E11R primer pairs. No fragments were amplified from any other fungus associated with plant disease (Supplementary Fig. S2). However, these primer .pairs were specific to V. nashicola as amplification of the expected fragments was successfully performed with DNA of all Korean isolates tested (Fig. 3).
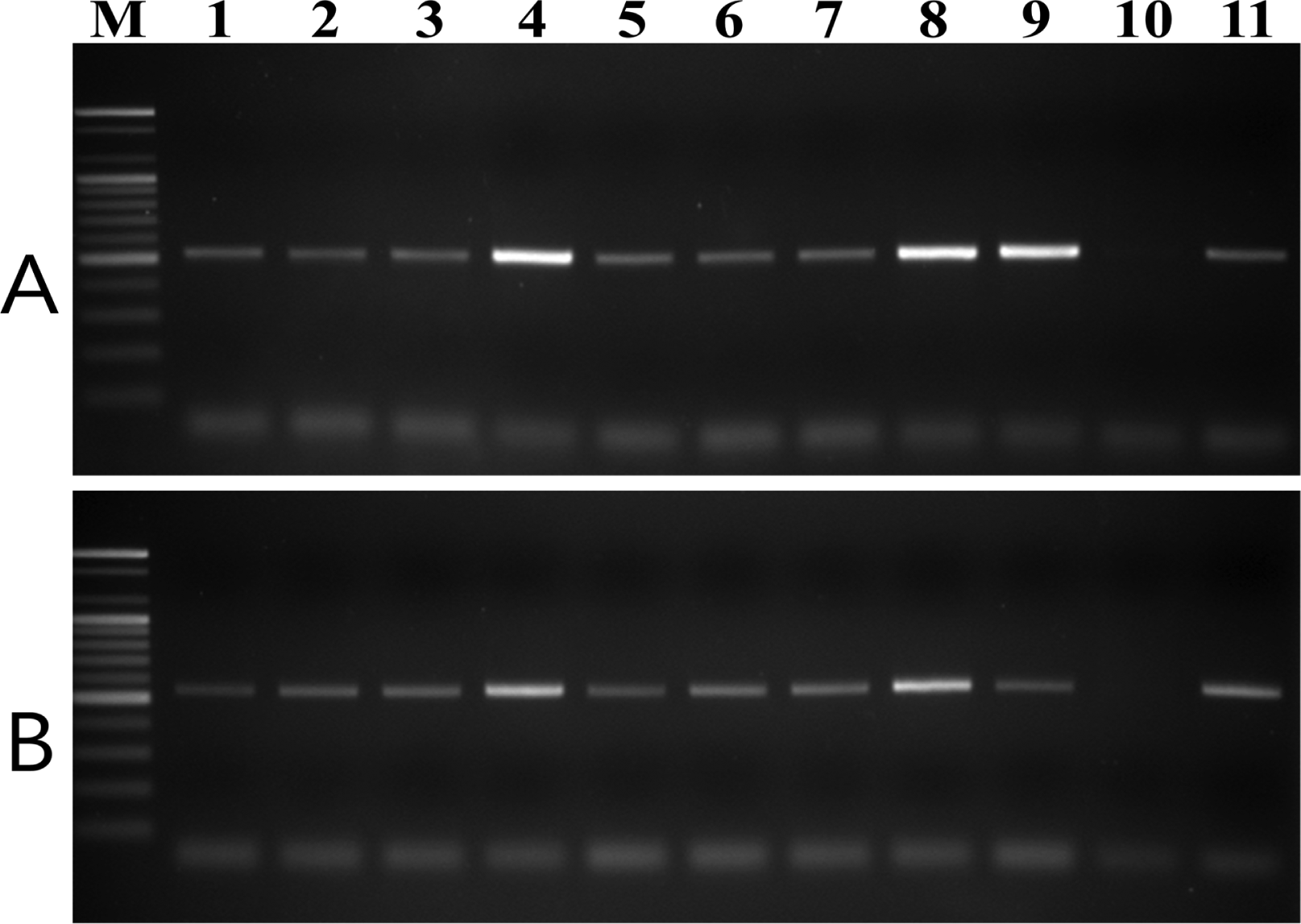
PCR amplification of genomic DNA of V. nashicola strains isolated in Korea with primer pairs (A) D12F/D12R and (B) E11F/E11R. Lanes 1–9 correspond to V. nashicola strain numbers 12–20 listed in Table 1; lane 10, V. pirina MAFF 615009; lane 11, V. nashicola MAFF 615023; lane M, 100 bp DNA size marker (Bioneer).
Evaluation of internal primer pair for the nested PCR
The internal primer pair Na11F (5′-CACCACCAGGTCAGATTTCG-3′)/Na11R (5′-GGTATTATCTACTCGATCAG-3′) amplified an expected 235-bp fragment only with V. nashicola DNA (Fig. 2C). No amplified product was generated from other Venturia species using this primer pair. Southern hybridization result also confirmed the specificity of this primer set to V. nashicola (Supplementary Fig. S1). When used as an internal primer pair for nested PCR, in combination with external primer pair E11F/E11R, Na11F/Na11R gave an amplification pattern identical to that of a single step PCR assay.
Sensitivity of nested PCR assay
In the nested PCR system, the external and internal primers amplified 525-bp and 235-bp DNA fragments, respectively, from all V. nashicola strains tested (data not shown). Sensitivity of the nested PCR system was determined by using different amounts of V. nashicola KCTC 6484 DNA as a template. The first PCR with primer pair E11F/E11R detected the specific fragment in 5 pg of V. nashicola DNA. Nested PCR assay using 1 μl of the first PCR product as a template was 100 times more sensitive than the single round PCR and could detect the expected DNA fragment in as little as 50 fg of V. nashicola DNA (Fig. 4).
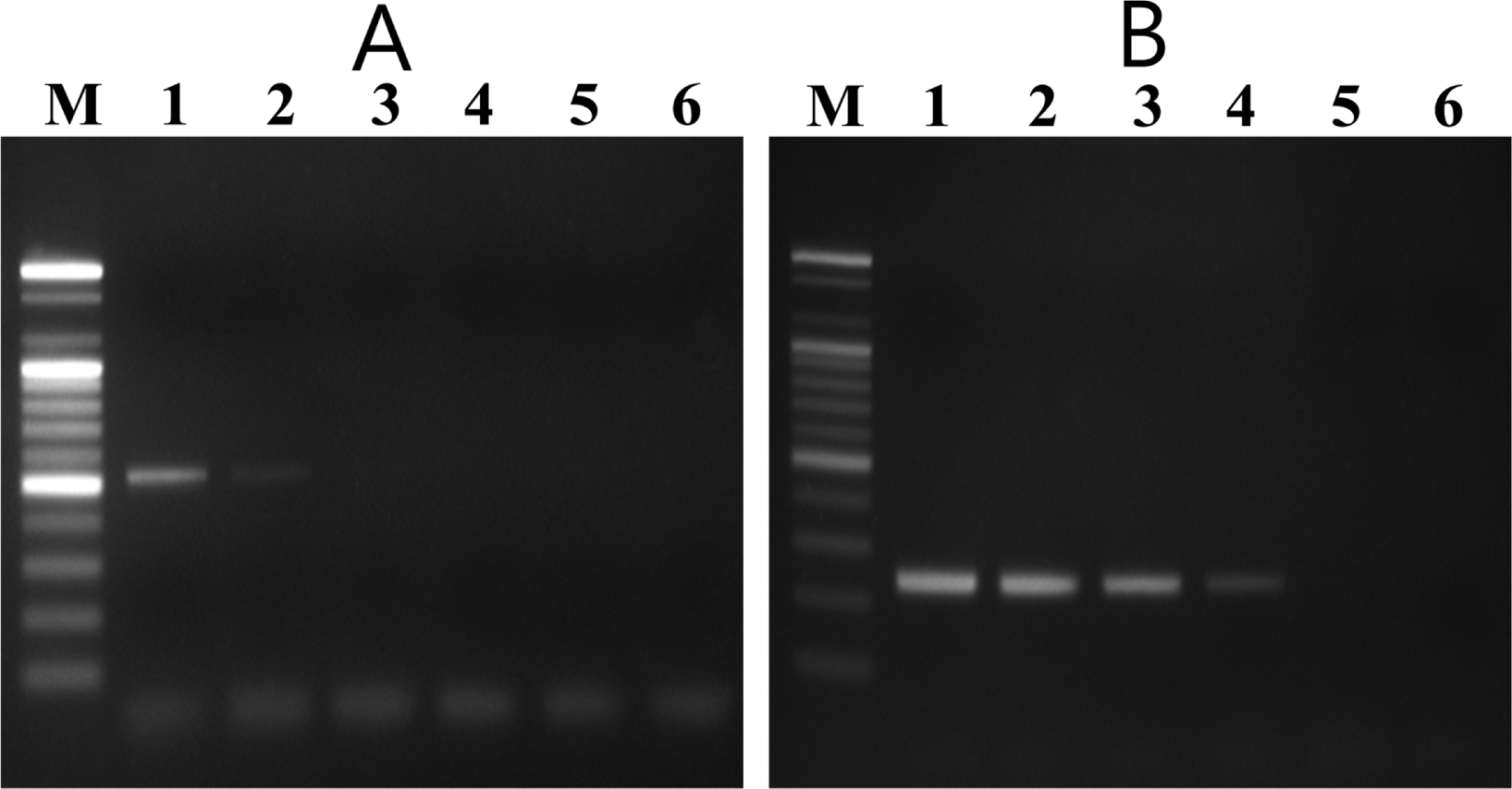
Sensitivity of V. nashicola primer pairs. (A) Single step PCR amplification with external primer pair E11F/E11R and (B) subsequent nested PCR with internal primer pair Na11F/Na11R for detection of V. nashicola KCTC 6484 DNA. Lane M, 100 bp DNA size marker (Bioneer); lanes 1–6: 50 pg, 5 pg, 500 fg, 50 fg, 5 fg, and 500 ag of genomic DNA of V. nashicola.
Discussion
V. nashicola, the causal agent of scab on Asian pear cultivars, is one of the most serious pear pathogens and is listed as a quarantine organism by many countries where Asian pear is poorly cultivated. It has been difficult to differentiate V. nashicola from V. pirina, the pathogen of European pear scab, as the two species are closely related. Recently, the phylogenetic relationship between V. nashicola and V. pirina was clarified on the basis of multiple nucleotide sequence analyses using ITS region of rDNA, β-tubuline and elongation factor 1α genes (Beck et al., 2005; Zhao et al., 2012).
V. nashicola and V. pirina can be distinguished by morphological characterization of the size of their conidia and ascospores. Conidia of V. nashicola are shorter than those of V. pirina, which have longer and wider ascospores (Ishii and Yanase, 2000). However, morphological differentiation of these two species is tedious, time consuming, and not suitable in practice. Morphological and biochemical characterizations of either fungus require its isolation in culture. However, isolation of Venturia species on selective media are subject to limitations since these pathogens grow slowly and could be masked by overgrowth of other faster growing fungi. For identification of V. nashicola, therefore, rapid and specific detection methods that can be applied to environmental samples are required.
Polymerase chain reaction has provided a sensitive, rapid, and reliable method for the identification and detection of plant pathogenic fungi (Martin et al., 2000). The sequences of the ITS1 and ITS2 spacer regions of fungal rDNA are variable and have potential to distinguish fungi at the species level. A PCR-based method using ITS sequence for the detection of V. nashicola was described previously by Le Cam et al. (2001). They aligned the complete ITS1 nucleotide sequences of V. nashicola, V. pirina and V. inaequalis and designed PCR primers specific to V. nashicola. The forward primer A126 was designed from a polymorphic region between V. nashicola and V. pirina, whereas the reverse primer was the “universal” ITS4 oligonucleotide from 28S rDNA (White et al., 1990). However, V. nashicola and V. pirina show only two base substitutions in the primer A126 region. The primer pair A126/ITS4, therefore, has the potential to amplify false positive band with DNA of V. pirina depending on the PCR conditions. We found that this PCR primer set produced a 391-bp band under recommended conditions with V. pirina when DNA hybridization was performed using the amplified fragment as a probe (data not shown).
In the present study, we designed two pairs of specific primers for V. nashicola using SCAR markers derived from RAPD analyses. As shown in Fig. 1, V. nashicola produced polymorphic and distinguishable RAPD band patterns comparable with its close relatives, V. pirina and V. inaequalis. RAPD markers were subsequently converted to SCAR markers by developing longer, and hence, more specific primers from RAPD sequences. After confirmation, the developed SCAR primer pairs, D12F/D12R and E11F/E11R, were validated with 76 additional Korean isolates. All Korean isolates tested produced expected fragments using both primer pairs.
Nested PCR was employed to increase the detection limit of the pathogen. In this study, we used the Na11F/Na11R pair as an internal primer of the second PCR. The specificity of this primer pair for V. nashicola was also confirmed by DNA hybridization as shown in Fig. 2C and supplementary Fig. S1. Therefore, three sets of PCR primers designed in this work can each be used as a primer for the first PCR to detect V. nashicola. Nested PCR employing V. nashicola-specific primers in both PCR rounds would satisfy the specificity and sensitive requirements for the monitoring applications. This approach shows high specificity and sensitivity for the detection of V. nashicola in a high background of other fungi and plant tissues.
In conclusion, the PCR primers designed in this work are sensitive and specific for the detection of V. nashicola. The detection method reported here could be used for the quarantine inspection of V. nashicola and the diagnosis of scab of Asian pears.
Acknowledgements
This work was supported by a grant “The Project for Export of Agricultural Products” from the Animal and Plant Quarantine Agency, Republic of Korea.