Discrimination and Detection of Erwinia amylovora and Erwinia pyrifoliae with a Single Primer Set
Article information

Abstract
Erwinia amylovora and Erwinia pyrifoliae cause fire blight and black-shoot blight, respectively, in apples and pears. E. pyrifoliae is less pathogenic and has a narrower host range than that of E. amylovora. Fire blight and black-shoot blight exhibit similar symptoms, making it difficult to distinguish one bacterial disease from the other. Molecular tools that differentiate fire blight from black-shoot blight could guide in the implementation of appropriate management strategies to control both diseases. In this study, a primer set was developed to detect and distinguish E. amylovora from E. pyrifoliae by conventional polymerase chain reaction (PCR). The primers produced amplicons of different sizes that were specific to each bacterial species. PCR products from E. amylovora and E. pyrifoliae cells at concentrations of 104 cfu/ml and 107 cfu/ml, respectively, were amplified, which demonstrated sufficient primer detection sensitivity. This primer set provides a simple molecular tool to distinguish between two types of bacterial diseases with similar symptoms.
Fire blight is a bacterial disease of plants in the Rosaceae family caused by Erwinia amylovora. The disease was first reported in New York in 1793 and has since spread to North America, Europe, Africa, Oceania, and Asia. Fire blight adversely impacts apple and pear production worldwide (Bonn and van der Zwet, 2000; Denning, 1794; Drenova et al., 2012; Fatmi et al., 2008; Park et al., 2016). E. amylovora infects the entire tree and young branches by moving systemically through the xylem after entering wounds and natural openings. Symptoms of fire blight are manifested as blights and necrotic lesions in flowers, leaves, and young stems. E. amylovora overwinters in cankers. When the temperature rises the following year, bacterial ooze forms, and the pathogen moves to other trees by rain, wind, insects, and other dispersal agents (Billing, 2011; Thomson, 2000).
Black-shoot blight is another bacterial disease in apples and pears. The disease is caused by Erwinia pyrifoliae. E. pyrifoliae was first reported in pear trees of South Korea in 1995 (Kim et al., 1999) and was also isolated from strawberries in the Netherlands in 2014 (Wenneker and Bergsma-Vlami, 2015). Virulence of E. pyrifoliae is lower than that of E. amylovora, and its host range is, for the most part, limited to pear and some varieties of apple (Kim et al., 2001). The trees infected by E. pyrifoliae show blackened blight and wilt in leaves and shoots, viscous exudates on the surface, and necrosis of the leaves, flowers, and immature fruits (Lee et al., 2020; Rhim et al., 1999).
Visually distinguishing the disease caused by E. amylovora from that caused by E. pyrifoliae is challenging because of their very similar symptoms of the infected trees (Kim et al., 2001). These two pathogens appeared in the South Korean provinces of Gangwon and Gyeonggi (Ham et al., 2020; Kim et al., 1999; Lee et al., 2020). Therefore, molecular detection methods provide valuable tools for distinguishing fire blight from black-shoot blight.
E. amylovora is usually detected by specific regions in the plasmid of pEA29 and chromosome by polymerase chain reaction (PCR). The pEA29 is indigenous and related to virulence in E. amylovora (Llop et al., 2006). It has been widely used because of its high sensitivity since it has more than one copy in the genome (Bereswill et al., 1995; Obradovic et al., 2007). But after the discovering the E. amylovora strains which do not possess the pEA29 (Llop et al., 2006), the primer designed from their chromosome has been widely used to detect E. amylovora strain. In particular, the primers targeting ams genes involved in exopolysaccharide (EPS) production were designed (Bereswill et al., 1995; Jones and Geider, 2001; Mohammadi et al., 2009), but the false-positive response was found from other bacteria (Powney et al., 2011).
E. pyrifoliae has minor occurrences except for Korea, so there is little information, and the developed primers are limited. The EP16A/EPIG2C primer amplifying the 16S rRNA and internal transcribed spacer region, and the CPS1/2C primer amplifying the cpsB and cpsC region, which encode EPS, were developed (Kim et al., 2001). But these primers were not verified to detect E. tasmaninasis and other closely related species that discovered later than at that time. In addition, the primers detecting both pathogens at once are not developed yet.
Here, comparative genomics was used to design a primer set from a single-copy gene to amplify specific DNA sequences from E. amylovora. This primer set also amplified E. pyrifoliae DNA but produced an amplicon that had a different size than that of E. amylovora. Sequencing the E. pyrifoliae amplicon confirmed the E. pyrifoliae genome binding site. In addition, testing the primers on genomic DNA (gDNA), bacterial suspension cells, and plant extracts revealed detection specificity and sensitivity of the primers. Therefore, this primer set is a valuable addition to molecular tools for the rapid and economical detection of E. amylovora and E. pyrifoliae. The primer set developed in this study is unique in that it can detect and distinguish one bacterial pathogen from the other with a single PCR.
Materials and Methods
Bacterial strains and cultural condition
Twenty-three bacterial species were collected for primer specificity assays (Table 1). Bacterial species from E. amylovora TS3128 and E. pyrifoliae YKB12327 were isolated from diseased pear or apple lesions in 2015. Other bacterial isolates were obtained from the Korean Agricultural Culture Collection (KACC, South Korea) or the Belgian Coordinated Collections of Microorganisms (BCCM). Each bacterial species was cultured in tryptic soy agar (TSA) at 27°C for 48 h, and single colonies were used for the assays.
Bacterial strains used in this study
Total genomic DNA extraction
To extract gDNA from bacteria, single colonies were inoculated on tryptic soy broth and incubated overnight at 27°C under shaking conditions at 150 rpm. Bacterial cultures were pelleted, and gDNA was extracted with the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). DNA concentration was measured with a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA) before performing conventional PCR.
Primer and PCR amplification
Specific primers were generated from the downloaded E. amylovora CFBP1430 genome sequence (GenBank accession no. GCA_000091565.1) and other closely related species from the National Center for Biotechnology Information (NCBI) GeneBank. The specific genes and primers were selected and designed from the E. amylovora CFBP1430 genome using the genome-wide comparison pipeline as previously described (Lang et al., 2010) with minor modifications.
For PCR amplification, gDNA template (1 μl), 10 mM of each forward and reverse primer, 10× premix (Cellsafe, Yongin, Korea), 1.25 unit of Taq polymerase, and 0.2 mM of dNTPs were combined to produce a 25 μl reaction mixture. PCR was performed with a C1000 Thermal Cycler PCR system (Bio-Rad, Hercules, CA, USA). The PCR conditions were as follows: pre-denaturation at 95°C for 5 min, 35 cycles of denaturation at 95°C for 30 s, annealing at 61°C for 30 s, extension at 72°C for 30 s, and a final extension at 72°C for 10 min. For gel electrophoresis, 5 μl of each PCR product was mixed with 1 μl of DNA loading star (Dyne Bio, Seongnam, Korea) and then loaded onto a 1% agarose gel. The gel was visualized with a UV transmission illuminator after electrophoresis.
Configuration of the primer binding site of E. pyrifoliae
To identify the RS24580-205 primer binding site on the E. pyrifoliae genome, the 705 bp amplicon from the E. pyrifoliae template was cloned to the pGEM-T Easy Vector (Promega) and transformed to Escherichia coli DH5α competent cells. The resulting plasmid DNA was purified with GeneAll Hybrid-Q Plasmid Rapidprep (GeneAll Biotechnology, Seoul, Korea) and sequenced (Bionics, Daejeon, Korea) with T7 and SP6 universal primers. The primer binding site was determined by comparing the insertion sequences of the vector with that of the E. pyrifoliae DSM12163 genome sequence (GenBank accession no. GCA_000026985.1).
Specificity and sensitivity assays
The gDNA of bacterial strains listed in Table 1 was used to test the specificity of the RS24580-205 primer set. DNA concentration was adjusted to 25 μg/μl, and 1 μl was used as a template. To test primer sensitivity, E. amylovora and E. pyrifoliae gDNA templates were serially diluted by 10-fold (i.e., from 5 ng/μl to 5 fg/μl). Primer sensitivity was also tested on bacterial cell suspension. Overnight cultures E. amylovora and E. pyrifoliae cells were adjusted to an optical density (OD) of 1.0 at 600 nm with a NanoDrop spectrophotometer and serially diluted by 10-fold. Cell suspensions (100 μl) were spread on TSA plates to determine E. amylovora and E. pyrifoliae cell density. Cell density of E. amylovora at an OD of 1.0 was 2.55 × 109 cfu/ml, while that of E. pyrifoliae was 1.25 × 109 cfu/ml. Diluted gDNA (1 μl) and cell suspensions were used for PCR. The detection limits were determined by examining the amplified target bands.
Detection of E. amylovora and E. pyrifoliae on inoculated apple shoots
Shoot segments (15–20 cm long) were collected from branches of 2-year-old apple trees (cultivar Fuji) and surface sterilized with 70% ethanol. Shoots were put into water-soaked floral foam (Oasis, Smithers-Oasis Korea, Cheonan, Korea) and inoculated with 10 μl E. amylovora or E. pyrifoliae cell suspensions adjusted to an OD600 of 0.1 (108 cfu/ml) with 200 μl sterilized tips (TipOne UltraPoint Graduated Tip, Starlab Ltd., Hamburg, Germany). Inoculated shoots were incubated in a growth chamber maintaining 27°C and 100% relative humidity, and disease symptoms were observed after 7 days. After sterilizing the surface of the shoots with 70% ethanol, lesions were cut into 5 × 5 mm pieces. Five lesions were ground in 1 ml sterile distilled water, and 200 μl of suspension was used to extract total DNA using the taco plant DNA/RNA extraction kit (GeneReach Biotechnology Corp., Taichung City, Taiwan) with nucleic acid automatic extraction system (taco, GeneReach Biotechnology Corp.).
Results and Discussion
Primer design, PCR detection, and specificity test
From the NCBI GenBank FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/), we identified unique chromosome loci of E. amylovora against the loci of all available Erwinia genomes using a BLASTN (E = 1e–1) search. By the Primer3 module (Rozen and Skalestsky, 2000), primers producing 95–310 bp of amplicons were designed from the unique E. amylovora loci. Using reverse-ePCR (n = 2, g = 1) (Schuler, 1997), predicted primer pairs were confirmed against all Erwinia genomes. Primers only for targeting the E. amylovora genome were selected. Finally, the forward primer, RS24580-205F (5′-CAC TGC GCC TGT TGT TCA-3′), and reverse primer, 205R (5′-ATG TAT CTG GTA GCC GGG TAA GTT-3′), were selected which encodes a hypothetical protein (WP_162010770.1) from the E. amylovora genome (Fig. 1). We also found that the RS24580-205 primers amplified not only 205 bp of PCR product from E. amylovora genome but also 751 bp from E. pyrifoliae (Table 1, Fig. 2, lanes 1–10). Other closely related bacterial species, such as Pectobacterium, Dickeya, and Pantoea sp., which were previously classified as Erwinia species, and five strains belonging to the Erwinia genus, were used to test the specificity of the RS24580-205 primers (Kado, 2006; Samson et al., 2005) (Table 1). No PCR products were amplified from other bacterial species with the RS24580-205 primer set (Table 1, Fig. 2, lanes 11–23).

Comparative analysis of Erwinia amylovora strains of specific binding sitez in hypothetical protein (WP_162010770.1) gene. (A) The BLASTN search of hypothetical protein gene from E. amylovora CFBP1430. (B) Comparison and alignment of primer binding sites among E. amylovora strains.
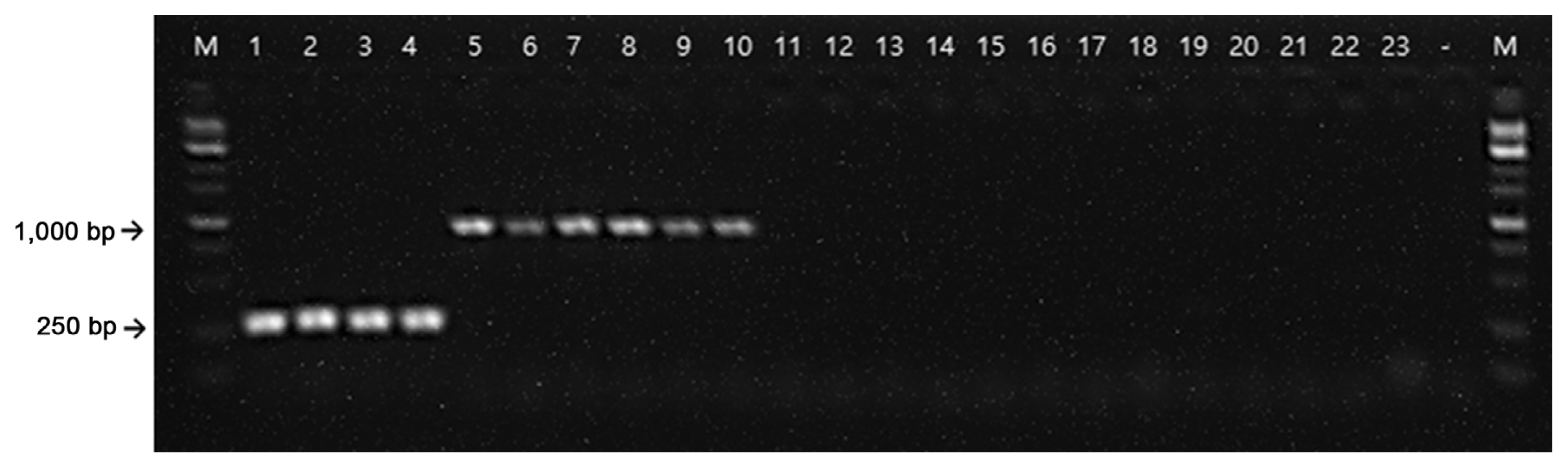
Polymerase chain reaction amplification of Erwinia amylovora and Erwinia pyrifoliae with the RS24580-205 primer set. M, 1 kb size marker; lanes 1–4, 25 ng E. amylovora gDNA; lanes 5–10, E. pyrifoliae gDNA; lanes 11–23, other Erwinia, Pectobacterium, Dickeya, and Pantoea species listed in Table 1; lane −, negative control.
In contrast to the RS24580-205 primers developed in this study, it was reported that A/B primers, which were designed to specifically amplify the sequences in the E. amylovora pEA29 plasmid at the amplicon size between 900 bp, produced non-specific bands by PCR. The detection of Erwinia herbicola (Pantoea agglomerans) and Erwinia carotovora sub-species atroseptica (Pectobacterium atrosepticum) by A/B primers did not produce 900 bp of amplicon but shorter than or above the 900 bp (Bereswill et al., 1992).
In conclusion, these results showed that in addition to the high specificity of RS24580-205 primers for E. amylovora and E. pyrifoliae, the generation of a different amplicon size specific for each bacterial species made it possible to distinguish one pathogen from the other using a single primer set (Fig. 2).
Amplification regions by RS24580-205F/R primer in E. pyrifoliae
The forward primer, RS24580-205F, bound to a ‘metal-dependent hydrolase (WP_012667018.1)’ sequence of the E. pyrifoliae DSM12163 genome, while the reverse primer, RS24580-205R, bound to a gene encoding a ‘patatin-like phospholipase family protein (WP_012667017.1)’ (Fig. 3). Patatin-like proteins are the storage proteins and have lipolytic enzyme activity that related to the plant–pathogen interactions (Banerji and Flieger, 2004). For the 18-mer RS24580-205F primer, 14-mer at the 3′ end was complementary to the target template, while 3-mer at the 5′ end was mismatched. In the 24-mer RS24580-205R primer, 12-mer at the 3′ end was complementary to the target template, while 4-mer at the 5′ end was mismatched. The binding features of the RS24580-205 primer set to the E. pyrifoliae templates resulted in a 751 bp amplicon. Although each primer did not bind perfectly to the E. pyrifoliae genome sequence, more than 12-mer at the 3′ end was complementary, which was sufficient to generate a PCR product.
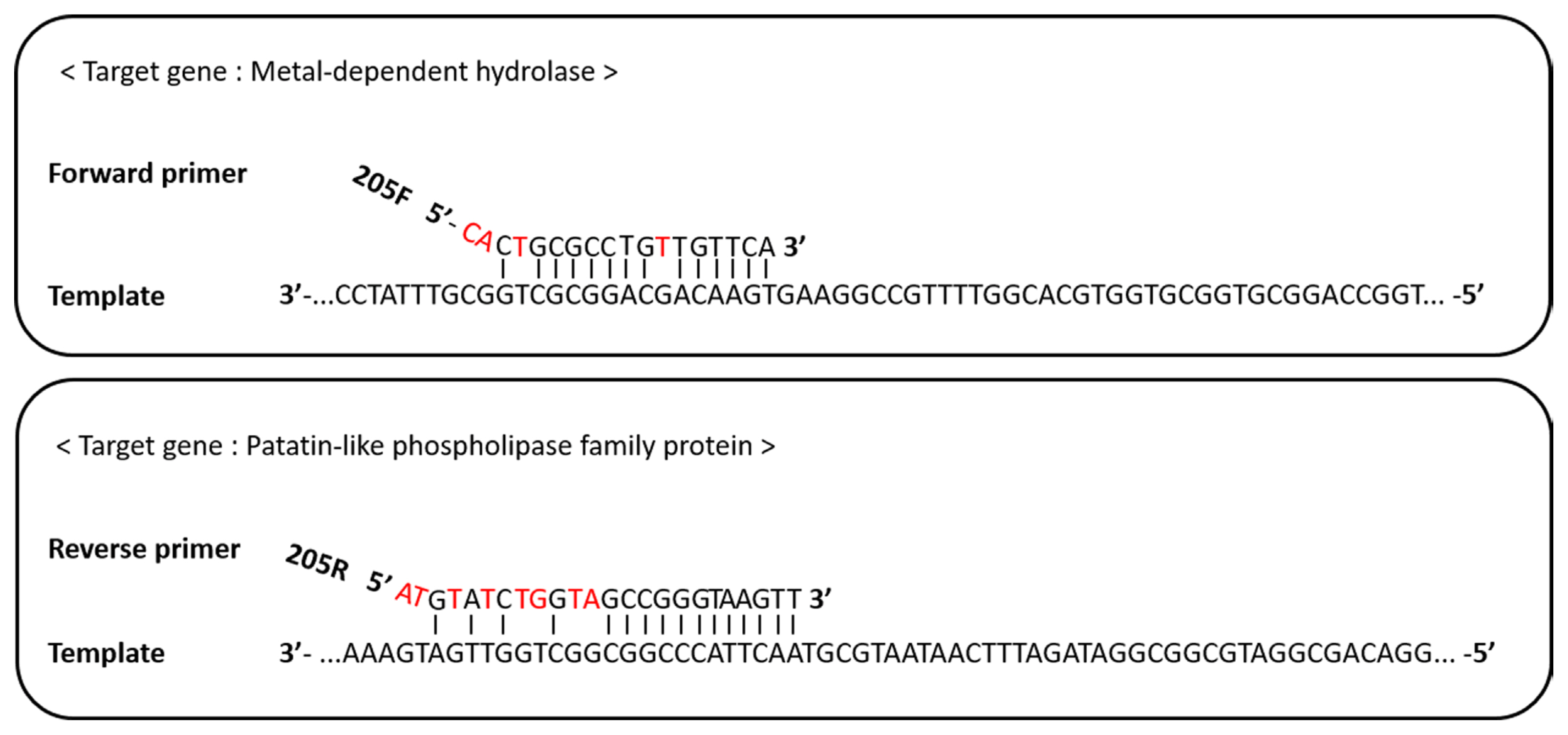
Binding site of the RS24580-205F/R primer to the Erwinia pyrifoliae genomic DNA template.
In general, mismatched bps at the 3′ end of the primer have a more adverse effect on PCR amplification than those at the 5′ end. If there are several bp mismatches at the 5′ end or the middle of the primer, target amplification can still occur, but efficiency is reduced (Ye et al., 2012). Also, 3-bp mismatches at the 5′ end of the forward primer cannot amplify the target template when its concentration is low (Sipos et al., 2007). The efficiency of RS24580-205F and RS24580-205R primers was reduced because of 4-mer and 8-mer bp mismatches, respectively, at the 5′ end. However, the 12-mer match at the 3′ end of the RS24580-205 primer set still allowed template amplification. The binding regions of the primers to E. pyrifoliae templates were compared to those of E. amylovora, E. billingiae, E. rhapontici, E. tasmaniensis, P. agglomerans, and P. dispersa (Supplementary Fig. 1). The binding sequence of the metal-dependent hydrolase gene to the RS24580-205F primer was the same for E. pyrifoliae, E. amylovora, and E. tasmaniensis. However, the binding sequence of the patatin-like phospholipase family protein gene to the RS24580-205R primer was unique to the E. pyrifoliae template, which led to the 751 bp amplicon. The ability of the RS24580-205 primer set to simultaneously detect E. amylovora and E. pyrifoliae was tested by mixing the gDNA or bacterial cells for a 1:1 ratio from both pathogens. Only the 205 bp amplicon from E. amylovora was identified, which indicated partial target detection of E. pyrifoliae when both pathogens were infected together.
Sensitivity tests of the designed primers
Sensitivity tests of RS24580-205 primers showed that in E. amylovora, gDNA was detected at concentrations as low as 500 fg, while that of E. pyrifoliae was 50 pg (Fig. 4A and B). The detection limit for E. amylovora gDNA was 100 times higher than that for E. pyrifoliae. In bacterial suspensions, the RS24580-205 primer detected E. amylovora template from cells at a concentration of 104 cfu/ml, which corresponded to 10 cfu/reaction or higher. On the other hand, the primer set only detected E. pyrifoliae template from cells at a concentration of 107 cfu/ml, which corresponded to 104 cfu/reaction (Fig. 4C and D).
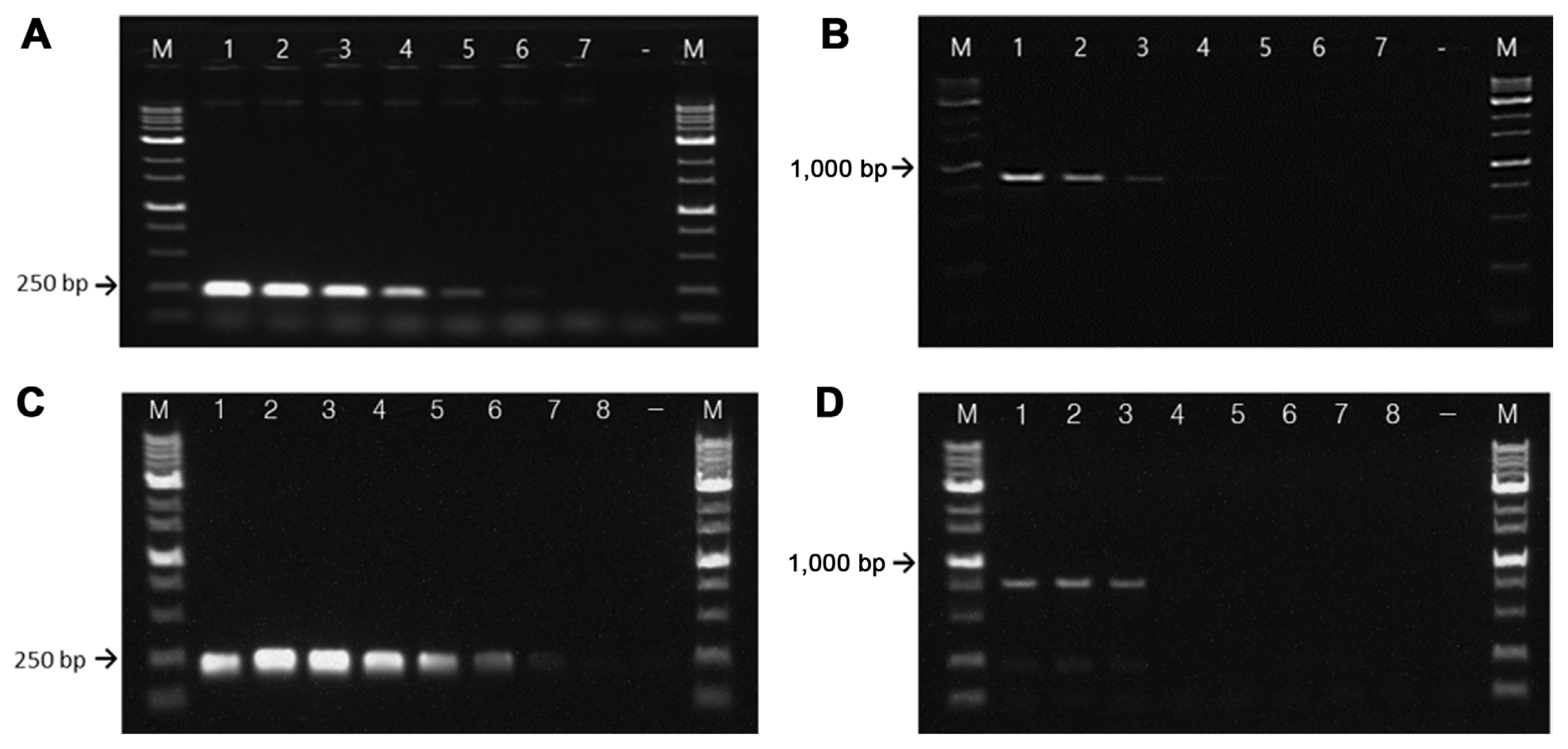
Sensitivity of the RS24580-205 primer set using serially diluted bacterial genomic DNA and suspension cells. Genomic DNA (gDNA) of Erwinia amylovora TS3128 (A) and E. pyrifoliae CP12327 (B). M, 1 kb size marker; lane 1, 5 ng gDNA; lane 2, 500 pg gDNA; lane 3, 50 pg gDNA; lane 4, 5 pg gDNA; lane 5, 500 fg gDNA; lane 6, 50 fg gDNA; lane 7, 5 fg gDNA; −, negative control. Bacterial cells of E. amylovora TS3128 (C) and E. pyrifoliae CP12327 (D). M, 1 kb size marker; lane 1, 109 cfu/ml; lane 2, 108 cfu/ml; lane 3, 107 cfu/ml; lane 4, 106 cfu/ml; lane 5, 105 cfu/ml; lane 6, 104 cfu/ml; lane 7, 103 cfu/ml; lane 8, 102 cfu/ml; −, negative control.
There are several primers developed to detect E. amylovora DNA sequences. For example, the A/B primer alluded to earlier, which targets the E. amylovora pEA29 plasmid, can detect 1 pg of E. amylovora gDNA or 1.9 × 103 cfu/ml of bacterial cells (Bereswill et al., 1992; Ivanović et al., 2019). The G1/G2 primers (also called pEA71 primers), which target an E. amylovora chromosomal sequence, have a detection limit of 10 cfu/reaction (Taylor et al., 2001). Another E. amylovora chromosome-targeted primer set, FER1/R, reproducibly shows a detection limit of 3 cfu/reaction (Obradovic et al., 2007). However, another report shows that the detection limit of FER1/R for bacterial cells is as low as 1.9 × 107 cfu/ml, which corresponds to 1.9 × 105 cfu/reaction (Ivanović et al., 2019). The RS24580-205 primer set developed in this study showed similar or higher sensitivity for E. amylovora detection than that of other primers, except for the A/B primer set. Nonetheless, A/B primers cannot detect E. amylovora strains lacking pEA29 (Llop et al., 2006). Also, there are several copies of pEA29 in the E. amylovora genome, which increases the sensitivity of the A/B primer set (Bereswill et al., 1995; Obradovic et al., 2007). By contrast, the RS24580-205 primer was designed to target a single-copy gene. Primers that target single-copy genes are beneficial for measuring PCR efficiency. When we use 10-fold serial dilution of target DNA to draw a calibration curve from the real-time PCR, single-copy gene makes the slope of −3.322 of regression equation at 100% of PCR efficiency. This means polymerase reaction of target DNA is in the exponential rate, which enables reliable quantification results (Ginzinger, 2002; Kralik and Ricchi, 2017). Furthermore, the PCR amplicon of E. amylovora from the primer is 205 bp, suitable for applying real-time PCR. Therefore, the RS24580-205 primer set can provide a new tool for quantitative studies of E. amylovora.
There are also primers for detecting E. pyrifoliae cells. For instance, the EP16A/EPIG2C primer set detects DNA from E. pyrifoliae bacterial cells at 20 cfu/reaction, which corresponds to 2 × 104 cfu/ml, while CPS1C/2R detects 200 cfu/reaction, which is about 2 × 105 cfu/ml (Kim et al., 2001). Moreover, the EpERF/R primer set, which targets enterobacterial repetitive intergenic consensus sequences, has a detection limit of 50 pg for gDNA and 104 cfu/ml for bacterial cells (Park et al., 2010). The sensitivity of these primers is higher than that of the RS24580-205 primer. Because they are only targeted for E. pyrifoliae, which enables more sensitive detection. Nonetheless, the fact that RS24580-205 primer set detects and discriminates E. amylovora and E. pyrifoliae at once is the greatest advantage of this primer.
Detection of pathogens from inoculated apple shoots
DNA from the lesions of apple shoots inoculated with bacterial suspensions (Fig. 5A) was amplified with the RS24580-205 primer set. The detection limit of the primers for plant extracts was 150 pg of total gDNA, which corresponded to 3.7 × 105 cfu/ml in E. amylovora (Fig. 5B), and 15 ng of total g DNA, which corresponded to 1.1 × 107 cfu/ml in E. pyrifoliae (Fig. 5C). The detection sensitivity of the RS24580-205 primers for DNA of plant extracts was 100 times higher in E. amylovora than in E. pyrifoliae. Also, the detection sensitivity of the primers for bacterial gDNA was 300 times higher than that for DNA from plant extracts. By contrast, A/B primers detect E. amylovora from DNA of plant extracts at 105–106 cfu/ml. G1/G2 and FER1-F/rgER2R detect plant DNA at 103–105 cfu/ml, while that of PEANT1/2 is 104–106 cfu/ml (European and Mediterranean Plant Protection Organization, 2013). On the other hand, EP16A/EPIG2c and CPS1/CPS2c primers, which target E. pyrifoliae, detect 103 cfu/ml of bacterial cells from extracts of diseased plant materials (Kim et al., 2001). The ERIC1R/ERIC2 primer set was reported to detect E. pyrifoliae from the total DNA of plant materials, inoculated with 106 cfu/ml of E. pyrifoliae (Park et al., 2010). The detection sensitivity of the RS24580-205 primer set from the E. amylovora infected plant materials showed similar levels with the previously developed primers. However, the detection of the primer set from the plant materials infected by E. pyrifoliae was less sensitive than the former studies.

Polymerase chain reaction amplification of bacterial isolates from diseased apple leaves using the RS24580-205 primer set. (A) Pathogen-inoculated apple shoots. SDW, inoculated with sterilized distilled water; EA, Erwinia amylovora TS3128; EP, Erwinia pyrifoliae CP12327. Red arrows point to inoculated sites. (B) Genomic DNA (gDNA) of E. amylovora TS3128-inoculated diseased apple shoots. M, 1 kb size marker; lane 1, 15 ng gDNA; lane 2, 1.5 ng gDNA; lane 3, 150 pg gDNA; lane 4, 15 pg gDNA; lane 5, 1.5 pg gDNA; lane 6, 150 fg gDNA; lane 7, 15 fg gDNA. (C) gDNA of E. pyrifoliae CP12327 inoculated diseased apple shoots. M, 1 kb size marker; lane 1, 15 ng gDNA; lane 2, 10 ng gDNA; lane 3, 5 ng gDNA; lane 4, 1 ng gDNA; lane 5, 500 pg gDNA; lane 6, 50 pg gDNA.
In summary, the RS24580-205 primer set described here detected templates of E. amylovora and E. pyrifoliae by conventional PCR. By binding to different regions of the E. amylovora and E. pyrifoliae genome, this primer set produced amplicons of 205 bp and 751 bp for E. amylovora and E. pyrifoliae, respectively, making it possible to distinguish one pathogen from the other with a single PCR reaction. The RS24580-205 primer set detected E. amylovora with high sensitivity. However, sensitivity for E. pyrifoliae was somewhat low because of primer mismatches at the 5′ end. Despite limitations in sensitivity, the ability of the RS24580-205 primer set to distinguish E. amylovora from E. pyrifoliae based on amplicon size is a valuable addition to the suite of molecular tools to combat these bacterial diseases through early detection.
Electronic Supplementary Material
Supplementary materials are available at The Plant Pathology Journal website (http://www.ppjonline.org/).
Acknowledgments
This work was supported by the “Cooperative Research Program for Agriculture Science and Technology Development” (Project No. PJ01421904), Rural Development Administration, Republic of Korea.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.