Culturing Simpler and Bacterial Wilt Suppressive Microbial Communities from Tomato Rhizosphere
Article information
Abstract
Plant phenotype is affected by a community of associated microorganisms which requires dissection of the functional fraction. In this study, we aimed to culture the functionally active fraction of an upland soil microbiome, which can suppress tomato bacterial wilt. The microbiome fraction (MF) from the rhizosphere of Hawaii 7996 treated with an upland soil or forest soil MF was successively cultured in a designed modified M9 (MM9) medium partially mimicking the nutrient composition of tomato root exudates. Bacterial cells were harvested to amplify V3 and V4 regions of 16S rRNA gene for QIIME based sequence analysis and were also treated to Hawaii 7996 prior to Ralstonia solanacearum inoculation. The disease progress indicated that the upland MM9 1st transfer suppressed the bacterial wilt. Community analysis revealed that species richness was declined by successive cultivation of the MF. The upland MM9 1st transfer harbored population of phylum Proteobacteria (98.12%), Bacteriodetes (0.69%), Firmicutes (0.51%), Actinobacteria (0.08%), unidentified (0.54%), Cyanobacteria (0.01%), FBP (0.001%), OD1 (0.001%), Acidobacteria (0.005%). The family Enterobacteriaceae of Proteobacteria was the dominant member (86.76%) of the total population of which genus Enterobacter composed 86.76% making it a potential candidate to suppress bacterial wilt. The results suggest that this mixed culture approach is feasible to harvest microorganisms which may function as biocontrol agents.
The mutualistic interactions between plants and microorganisms culminate in a reciprocally beneficial environment (Bulgarelli et al., 2013, Turner et al., 2013). Plants harbor an enormous number of microorganisms associated with them, and such associations are kept by investing a sizeable amount of energy in the form of rhizodeposits, which influence the health and growth of these host plants (Mendes et al., 2011, Philippot et al., 2013). These plant-associated microbial communities are also known to provide defense against plant pathogens (Balint-Kurti et al., 2010; Latz et al., 2012; van Elsas et al., 2012).
Plant roots are known to secrete root exudates, the compounds and signaling molecules, which not only provide protection against antagonist organisms but also recruit beneficial microorganisms (Bais et al., 2006; Kwak et al., 2018). The root exudates are composed of organic acids, amino acids and phenolics, and play an important role to establish plant-specific microbial communities in the rhizosphere (Sasse et al., 2018). Rhizosphere microbial communities are the most important player in the rhizosphere to make nutrients available to plants, produce phytohormores, degrade toxic compounds and suppress the soil-borne pathogens (Bais et al., 2006). It has been reported that the intricate and high microbial diversity in the soil and rhizosphere render it difficult to illustrate the functions of the microbial organization (Berendsen et al., 2012).
Various studies have been conducted by establishing model host with simplified and defined microbial communities to dissect the interactions between the host plant and defined communities (Bai et al., 2015; Bodenhausen et al., 2014; Lebeis et al., 2015). A study by Bodenhausen et al. (2014) has used a synthetic community approach to reveal that genotype of plants dictates the microbiota structure. The synthetic community approach in gnotobiotic systems makes it possible to take reductionist approaches to understand the role of community function as well as individual microorganisms (Vorholt et al., 2017). However, if one can culture microbial community per se, it would better resemble the natural microbiota to understand the role of the microbial community under defined in vivo condition. Therefore, this study, focus on reproducing a simplified and representative microbial community using the microbial mixed culture from the rhizosphere making plants either disease suppressive or disease conducive.
In this study we aimed to; (i) attempt to culture the major portion of the tomato rhizosphere microbial community per se as a mixed culture; (ii) to culture the functionally active fraction of an upland soil microbiome, capable of conferring quantitative resistance against tomato bacterial wilt caused by Ralstonia solanacearum. To do this, we used soil slurries (herein we coined terminology microbial fraction, MF) from two different soils. One of the soils was from upland soil that tomato plants have been cultivated, and the other soil was forest soil. Simply, soil slurries (i.e. MF) prepared from two different soils has been treated to tomato seedlings and the rhizosphere soils were harvested to subject microbiota cultivation.
The challenge to detail out the microbial community assemblage and function is directly proportional to the complexity of such microbial community. However, the microbial community harvested from our designed media, modified M9, is comparatively simpler than the community harvested from the tomato rhizosphere. Here, this study showed that our simplified mixed culture could define microbial alliance for bacterial wilt suppression in tomato and make it even possible to isolate microorganisms with biocontrol potential.
Materials and Methods
Plant material, growth conditions, pathogen culture and inoculum preparation
The plant used in this study was tomato (Solanum lycopersicum cv. Hawaii 7996). Prior to use, seeds were surface sterilized by vortexing in 70% EtOH added to 0.1% Triton X-100 for 1 min and 10% NaOCl added to 0.1% Triton X-100 for 15 min followed by thorough washing with sterilized water and drying in laminar flow hood. Both germination and plant growth conditions followed 10 h/14 h light/dark regime at 28°C. Seven-days-old seedlings were transferred to sterilized nursery soil (Punong Co., Ltd, Korea) and treated with microbiome fraction so that each gram of soil will receive ca 1 × 109 bacterial cells. Control plants were treated with 2.5 mM MES buffer (pH 5.7, adjusted by KOH) only. Plants were grown for 3 weeks prior to inoculation with R. solanacearum SL341 (race 1, phylotype I). The R. solanacearum SL341 cells suspension in sterilized water was adjusted to 1 × 108 cfu/ml (OD600 = 0.3) and was poured onto soil of unwounded plants at a final concentration of 1 × 107 cfu/g of soil. Bacterial wilt disease incidence was scored through 14 days. The disease incidence was rated using the 0–4 scale: 0, no wilting; 1, 1–25% leaves wilted; 2, 25–50% leaves wilted; 3, 51–75% leaves wilted; 4, 76–100% leaves wilted (Roberts et al., 1988).
Modified M9 (MM9) culture media
We designed this medium with an aim to mimic the root exudate chemical composition described by Kamilova et al. (2006). The minimal media M9 was supplemented with carbon sources, organic acids and trace elements to culture rhizosphere MF (Supplementary Table 1). Although the composition of root exudates was described to contain succinic acid, this organic acid was replaced by glutamic acid; an important amino acid for all living organisms, involved in various metabolic processes such as protein synthesis, glycolysis, gluconeogenesis and the citric acid cycle.
Microbiome fraction (MF) separation and treatment
MF separation was performed by two step centrifugation. In brief, for MF separation, top layer (5–10 cm) of upland bulk soil collected from Dong- A University farm in Gimhae and forest bulk soil collected from Seunghak Mountain were used. Bulk soil was added to 2.5 mM MES monohydrate buffer and incubated in an orbital shaker at 30°C, 200 rpm for 30 min. The soil suspension was centrifuged at 500 rpm for 5 min to remove soil particles and to collect the supernatant. The supernatant was then centrifuged at 8,000 rpm for 15 min to collect microbial pellets. The pellet was re-suspended in 2.5 mM MES monohydrate buffer (pH 5.7) to have final soil MF. Germinated seedlings planted were treated with the re-suspended MF so that each gram of soil receives 1 × 109 cfu/g. The plants were grown until 3 weeks.
Bacterial population cultured from rhizosphere MF in MM9 media
Rhizosphere soil of each 3 weeks old upland/forest MF treated Hawaii 7996 plants (n = 5) was immersed in 5 ml of 2.5 mM MES monohydrate (pH 5.7) and sonicated at 135 W for 5 min (Branson 5500DTH, Danbury, USA) with subsequent removal of the roots. The separated MF suspension was centrifuged at 500 rpm for 5 min, the supernatant was collected and subjected to high speed centrifugation at 8,000 rpm for 15 min to discard the supernatant and dissolving the pellet in 5 ml of 2.5 mM MES buffer. The MF of individual treatments from each of the 5 plants was pooled and transferred to 500 ml MM9 culture media in a 100 fold dilution. The media was incubated at 30°C, 100 rpm in rubber sealed flasks until the cell densities, accessed by measuring OD at 600 nm (DU® 730 Beckman Coulter® UV/Vis spectrophotometer), were 0.2–0.3. Prior to harvesting bacterial cells 5 ml of this cultured broth was transferred to a fresh batch of MM9 media (MM9 1st Tra) in a 100 × dilution and incubated until OD at 600 nm reached 0.2–0.3. The bacterial cells from each cultured broth were harvested by centrifugation at 8000 rpm for 15 min. The harvested bacterial cells were processed for; community analysis and re-suspended to treat 7-days-old seedlings for virulence assay. The pellet was suspended in 2.5 mM MES monohydrate buffer; 20 ml of this bacterial suspension was treated to a total of 30 seedlings per treatment. The plants were grown for a period of three weeks prior to inoculation with R. solanacearum SL341. Overall procedures for the mixed culture and subsequent virulence assays were presented in the schematic representation of experiment (Supplementary Fig. 1).
DNA extraction, 16S rRNA gene amplification and sequencing
The bulk soil from the 3 weeks old plants was manually removed to separate the rhizosphere soil, soil attached at soil-root interface. For each individual plant roots with rhizosphere soil attached were immersed in 50 ml falcon tubes, each containing 5 ml of 2.5 mM MES buffer (pH 5.7) and sonication was performed at 135 W for 5 min (Branson 5500DTH, Danbury, USA). After sonication, this 5 ml of soil suspension was processed for DNA extraction. To harvest bacterial DNA from mixed culture, the bacterial pellets from the MM9 media culture at specified OD were harvested by taking 1 mL of the culture (n = 5). The samples were subjected to centrifugation at 13,000 rpm for 5 min to collect bacterial pellets. The supernatant was discarded, and pellets were subjected to DNA extraction as described below.
DNA from rhizosphere soil and bacterial pellets harvested from MM9 media was extracted (n = 5) using FastDNA® Spin kit for soil (MP Biomedicals, USA) according to manufacturer’s protocol. The extracted DNA was used to amplify the hypervariable region V3–V4 of 16S rRNA gene using primer pair 341F-805R (Forward Primer 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG 3′; Reverse Primer = 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC 3′) (Herlemann et al., 2011) with added Illumina adapter overhang nucleotide sequences indicated by bold and underlined font. PCR amplification was performed using 2.5 μl microbial genomic DNA (5 ng/μl), 5 μl forward primer (1 μM), 5 μl reverse primer (1 μM), and 12.5 μl 2x KAPA HiFi HotStart Ready Mix PCR kit (Kapa Biosystems) at 95°C initial denaturation for 3 min, followed by 25 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 5 min. Reactions were cleaned up with Agencourt® AMPure® XP beads (Beckman Coulter Genomics) following previously described protocol (Zheng et al., 2015). Sequencing was performed on an Illumina MiSeq platform (NICEM, Republic of Korea).
Sequence analysis
The Illumina generated paired-end reads were merged using PANDAseq software (Masella et al., 2012). The data set was analyzed using Quantitative Insights Into Microbial Ecology (QIIME), an open-source software pipeline. For quality control sequences with phred quality (Q) score > 30 were removed, Forward reads truncated at 280 and Reverse at 200 were also excluded from the data set. From a total number of 6,671,809 sequence reads across all samples 2,312,751 number of reads were obtained after files were filtered, the total number of sequence reads before and after quality filtering are presented in Supplementary Table 2. Dereplication was performed using VSEARCH in QIIME, 1,482/3,301 chimeras found (44.9%) were removed and a total of 114,637 Operational Taxonomic Units (OTUs) were picked based on 97% similarity within reads using USEARCH (UPARSE). OTU taxonomic assignment and generation of an OTU table was performed using the Ribosomal Database Project and the USEARCH pipeline version 5.2.236, respectively (Edgar, 2013).
Diversity analysis
Bacterial alpha-diversity (Shannon diversity index and number of observed OTUs) was calculated from the rarefied OTU table using the QIIME script alpha_diversity.py to make even depths. Beta-diversity (Bray-Curtis dissimilarity measures) was calculated with a normalized OTU table employing CSS method with QIIME script CSS function normalize_table.py. Relative abundance analysis was performed with normalized OTUs. Individual OTUs in each sample were normalized by dividing the total counts of all OTUs within that sample, indicating relative abundance (RA) expressed as the taxonomy composition of each sample.
Isolation and identification of abundant OTU representing family Enterobacteriaceae
For isolating the bacterial strain representing OTU0, the glycerol stocks of the culture medium from upland MM9 1st Tra pellets were used. The pellets were resuspended in 2.5 mM MES buffer and serially diluted. The serial dilutions were spread on five different media types; TSA, R2A, Marine agar, LB and NB agar. From every media type the most abundant colonies were picked, purified and used for colony PCR amplification using the 16S rRNA V3 and V4 region using 341F-805R primers (Herlemann et al., 2011). The OUT0 representing family Enterobacteriaceae was found to be most abundant. DNA sequencing was conducted at the DNA sequencing facility of MacroGen (Seoul, Korea). The nucleotide sequence of the colony was identified by comparing with sequences in EzTaxon database (https://www.ezbiocloud.net).
Antagonistic activity of Enterobacteriaceae strain against R. solanacearum
The antagonistic activity of OTU0 of Enterobacteriaceae, was investigated against R. solanacearum both in vitro and in vivo. The ability of the OTU0 to suppress bacterial wilt disease was assessed using Hawaii 7996 planted in sterilized nursery soil and in non-sterilized nursery soil. The surface sterilized seeds of Hawaii 7996 were germinated for 7 days before transferring to soil. The plants were grown for two weeks and treated with the isolated strain at a final concentration of ca. 5.5 × 106 cfu/g of soil, corresponding to 0.1 OD600, suspended in PBS buffer (pH 7.4). This concentration was selected based on the original concentration of the OTU0 in the rhizosphere of Hawaii 7996 under upland MF treatment. The antagonistic activity of the isolate against R. solanacearum was assessed by soft agar (containing 0.6% agar) overlay method. Two hundred and fifty microliters of R. solanacearum suspension at 0.3 OD600, corresponding to 1 × 108 cell/ml, was added to 5 ml of CPG soft agar and poured onto a regular CPG agar plate. Prior to dropping 5 μl of bacterial cell suspension onto the agar the soft agar was allowed to cool and set. The antagonistic activity was observed by checking the clear zone around bacterial colony. For control, CPG soft agar containing only R. solanacearum and CPG soft agar without R. solanacearum onto which only bacterial cell suspensions were dropped were also grown.
Statistical analysis
All data analysis and visualization were run in R program (version 3.3.2) using Vegan, Phyloseq (McMurdie and Holmes, 2013) and ggplot2 packages. The bacterial wilt incidence was analyzed for significant difference among treatments by Repeated measures ANOVA on the data of wilted leaf percentage of each plant assessed for 14 days. For gauging normality, Shannon diversity indices of alpha diversity were used for Shapiro-Wilk test which subjected to Wilcoxon rank sum test. The variation among the bacterial communities harvested from the samples was observed by ANOVA based on distance matrices (ADONIS) test. Relative abundance of OTUs per sample was also subjected to Wilcoxon rank sum test for determining statistical significance.
Data deposition
The next-generation sequencing data of the 16S rRNA gene survey reported in this paper have been deposited in the GenBank database (BioProject ID no. PRJNA553490, accession nos. SRR9659330-SRR9659354).
Results
Bacterial wilt disease incidence
The efficacy of the cultured bacterial communities harvested from MM9 media inoculated with rhizosphere MF of upland and forest soils was investigated by assessing the bacterial wilt disease incidence on resistant tomato cultivar Hawaii 7996 (Fig. 1). Significant difference was observed by repeated measures ANOVA between control and upland MM9 (P = 0.001863), control and upland MM9 1st Tra (P = 0.0007244), forest MM9 and upland MM9 (P = 0.03541). No significant difference was found between upland MM9 and upland MM9 1st Tra (P = 0.7916472) control and forest MM9 (P = 0.3223), control and forest MM9 1st Tra (P = 0.06039), forest MM9 and forest MM9 1st Tra (P = 0.28440), forest MM9 and upland MM9 1st Tra (P = 0.07237), forest MM9 1st Tra and upland MM9 (P = 0.3079) and forest MM9 1st Tra and upland MM9 1st Tra (P = 0.4591). The bacterial wilt disease index (DI) scores indicated a statistically significant difference between plants treated with cultured microbial fraction harvested from media inoculated with upland rhizosphere MF and mock treated plants which is in line with the previous results (Choi, 2017) revealing the disease suppressive capacity of upland MF.
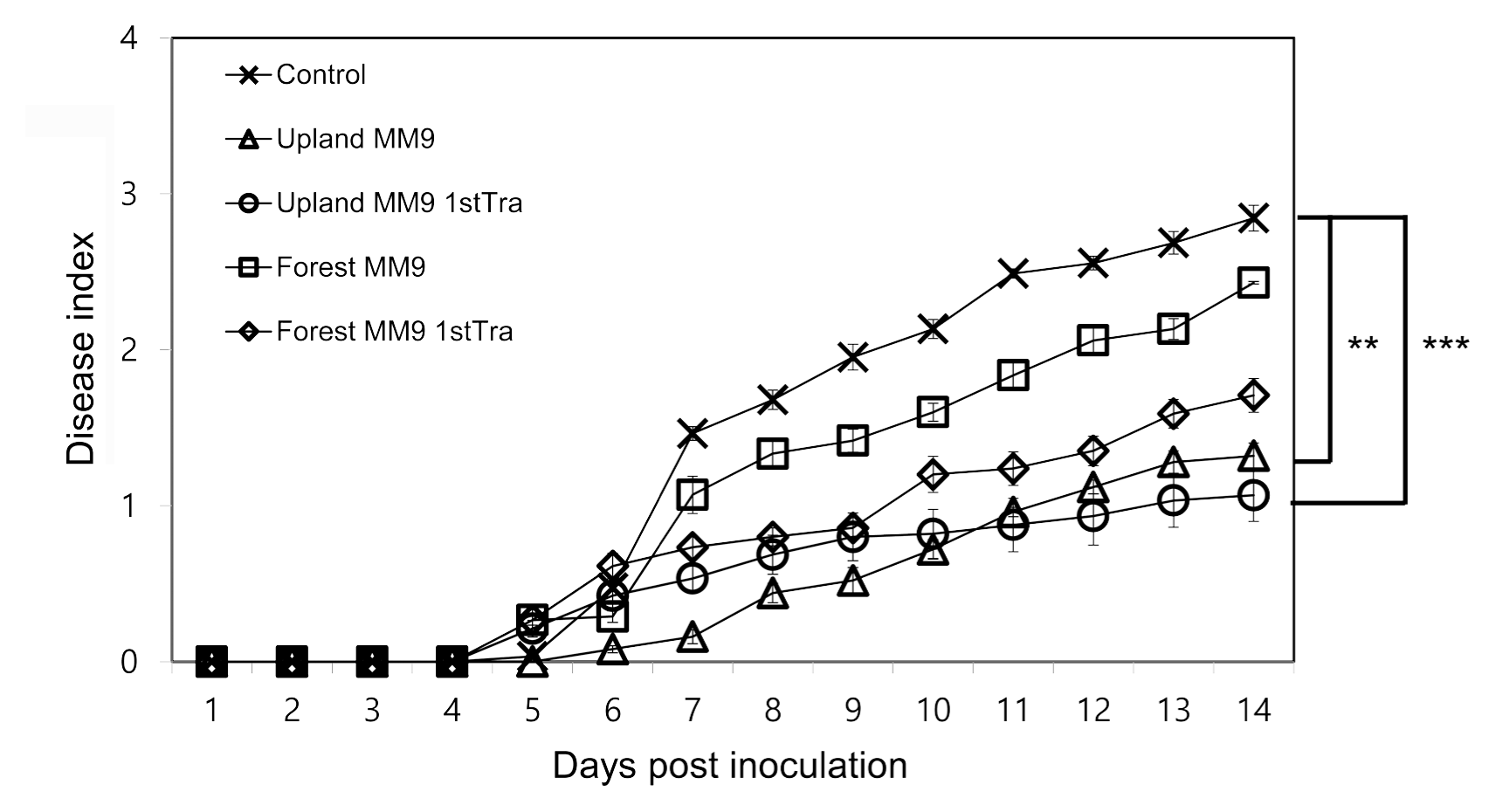
Disease progress of R. solanacearum strains SL341 on tomato. Thirty 21-days-old tomato plants (cv. Hawaii 7996) treated with bacterial cells harvested from inoculated MM9 culture media, control plants treated with 2.5 mM MES (pH 5.7) were inoculated by R. solanacearum SL341 at a final concentration of 1 × 107 cfu/g soil. Plants were rated for disease index through 14 days; vertical bars represent the standard error. Significant difference was observed by repeated measures ANOVA. P values are indicated by asterisks < 0.01 = **, < 0.001=***.
Variation among the microbial communities of samples
To view what level of variation in terms of community population dynamics existed among samples, measurements of variation were performed. Measurement of alpha diversity for observed species and Shannon diversity index (Fig. 2) ran on Wilcoxon signed-rank test. Significant difference for comparison between upland rhizosphere and upland MM9 (P = 0.006161), upland MM9 and upland MM9 1st Tra (P = 0.01965), upland rhizosphere and upland MM9 1st Tra (P = 0.001354), forest rhizosphere and forest MM9 (P = 0.01882) and forest rhizosphere and forest MM9 1st Tra (P = 0.009524) for observed species indices. In case of Shannon diversity index scores significant difference between upland rhizosphere and upland MM9 (P = 0.002876), upland MM9 and upland MM9 1st Tra (P = 0.01587), upland rhizosphere and upland MM9 1st Tra (P = 0.007937), forest rhizosphere and forest MM9 (P = 0.003322) and forest rhizosphere and forest MM9 1st Tra (P = 0.001778) were observed (Supplementary Table 3). Further, beta diversity was calculated by ADONIS which revealed significant variation (P = 0.001) among samples the R2 value equals 0.5283. Visualization was performed by using Non-metric multidimensional scaling (NMDS) analysis (Fig. 3).
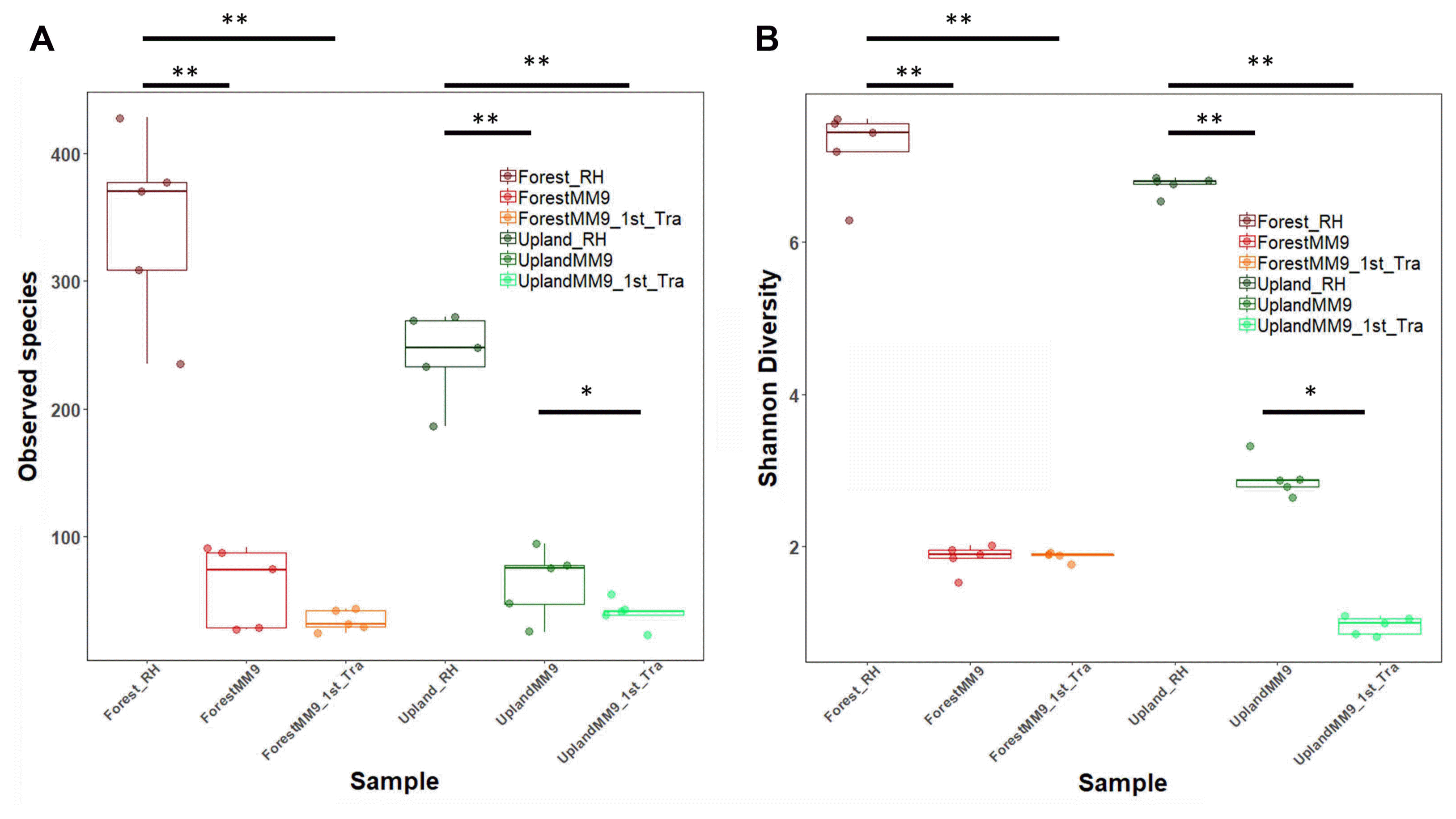
Bacterial community alpha diversity measured by observed species (A) and Shannon diversity index (B) from the rhizosphere MF of upland, forest MF treated Hawaii 7996 and their respective communities harvested from the cultures and subsequent cultures in MM9 media. The upland rhizosphere sample is abbreviated Upland_Rhi, forest rhizosphere sample as Forest_Rhi, bacterial mixed culture in MM9 inoculated with upland rhizosphere MF is abbreviated as UplandMM9, and bacterial mixed culture in MM9 transferred from UplandMM9 is abbreviated as UplandMM9_1st_Tra. The significance observed by Wilcoxon rank sum test are indicated by asterisks < 0.05 = *, < 0.01 = **.
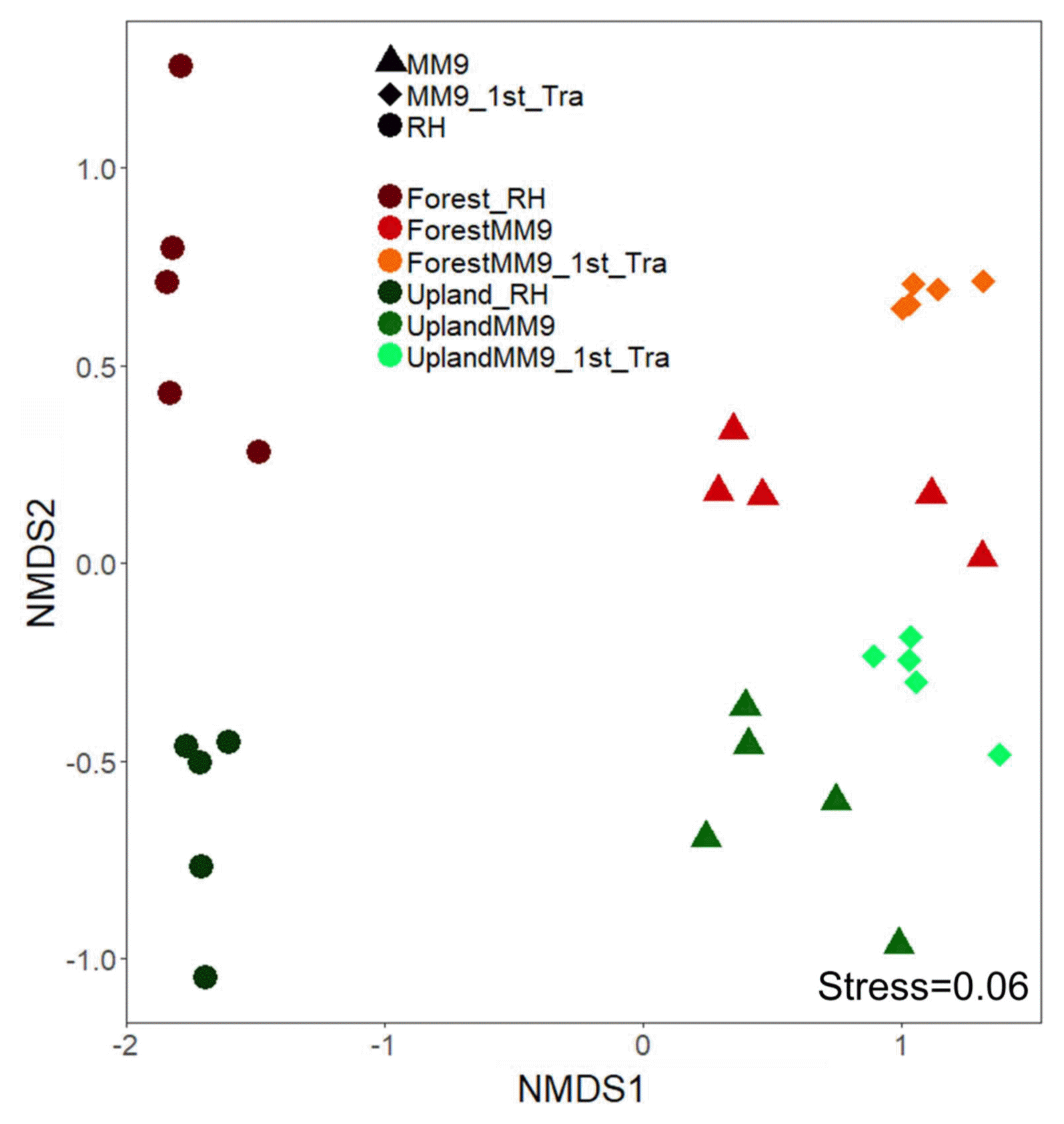
Non-metric multidimensional scaling (NMDS) plot of Bray-Curtis dissimilarity on microbial communities harvested from the rhizosphere MF of upland and forest MF treated Hawaii 7996 and their respective communities harvested from the cultures and subsequent cultures in MM9 media. NMDS plot stress value equals 0.06. Permutational multivariate analysis of variance (PERMANOVA) showed significant difference (P = 0.001) with R2 value 0.5283. The upland rhizosphere sample is abbreviated Upland_Rhi, forest rhizosphere sample as Forest_Rhi, bacterial mixed culture in MM9 inoculated with upland rhizosphere MF is abbreviated as UplandMM9, and bacterial mixed culture in MM9 transferred from UplandMM9 is abbreviated as UplandMM9_1st_Tra.
Mixed culture approach resulted in reproducible and simplified bacterial communities capable of conferring disease suppression
Mixed culture approach resulted in culturing a relatively simple and less diverse bacterial community compared to the original rhizosphere MF. The MM9 media was hence, capable to support the growth of the rhizosphere MF’s bacterial community. The culturing of rhizosphere MF in MM9 resulted in the detection of 50.64% of the original bacterial composition at family level in the upland MM9, 31.16% in the MM9 1st Tra, 52.4% in forest MM9 and 28.04% in forest MM9 1st Tra (Supplementary Table 4). This is not an indicative of the population of each family but rather the ability of MM9 medium to support growth, resulting in less diverse and simpler communities.
Population dynamics of upland and forest rhizosphere MF cultured in MM9 media at phylum level
The community composition of the major phyla (Fig. 4 and Supplementary Table 5) in the rhizosphere of the upland MF treated Hawaii 7996 plants was; Proteobacteria (43.50%), Bacteroidetes (15.38%), Firmicutes (14.25%), Planctomycetes (6.14%), Verrucomicrobia (6.02%), Actinobacteria (3.53%), TM7 (2.97%), and Acidobacteria (0.69%). The culturing of upland MF from rhizosphere in MM9 resulted in a population composition harboring; Proteobacteria (84.98%), Bacteroidetes (4.86%), Firmicutes (8.97%), Planctomycetes (0.07%), Verrucomicrobia (0.01%), Actinobacteria (0.35%), and Acidobacteria (0.02%). A subsequent transfer (i.e. second transfer) of the cultured upland MM9 broth media into a fresh batch resulted in a further change, stated as follows; Proteobacteria (98.12%), Bacteroidetes (0.69%), Firmicutes (0.51%), Planctomycetes (0.006%), Actinobacteria (0.08%), and Acidobacteria (0.004%).
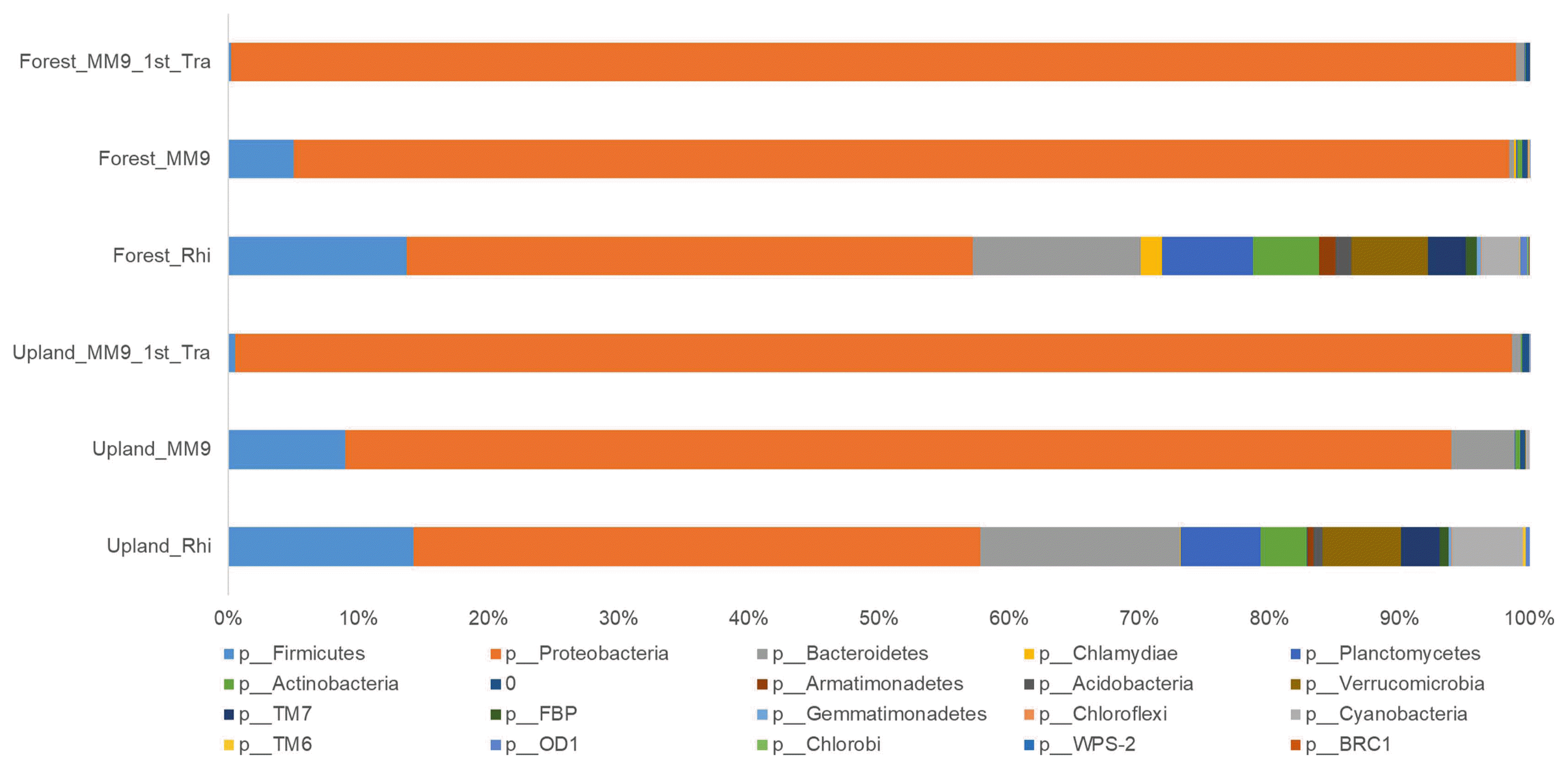
Relative abundance (%) of bacterial OTUs at the phylum level from tomato rhizosphere, bacterial mixed culture in MM9 inoculated with rhizosphere MF (MM9) and bacterial mixed culture in MM9 transferred from MM9 (MM9 1st Tra). The x-axis represents the relative abundance (%) of each phylum. The y-axis represents the samples. The color box below the figure represents the phyla.
Members from phyla Verrucomicrobia, TM7, Armatimonadetes, OD1, Gemmatimonadetes, TM6, Chlamydiae and Chloroflexi were not detected in the upalnd MM9 1st Tra culture. The phylum OD1 was undetectable in the upland MM9 culture but it was present within a detectable range in the upland MM9 1st Tra culture.
As for the rhizosphere of forest MF treated Hawaii 7996 the composition (Fig. 4 and Supplementary Table 5) of the population was; Proteobacteria (43.50%), Bacteroidetes (12.92%), Firmicutes (13.71%), Planctomycetes (6.97%), Verrucomicrobia (5.89%), Cyanobacteria (2.86%), Actinobacteria (5.10%), TM7 (2.92%), and Acidobacteria (1.17%). The shift in the composition compared to that of rhizosphere after culturing in MM9 for forest MF was; Proteobacteria (93.39%), Bacteriodetes (0.37%), Firmicutes (5.03%), Planctomycetes (0.14%), Verrucomicrobia (0.006%), Actinobacteria (0.34%), and Acidobacteria (0.05%). This harbored community when transferred from the forest MM9 culture to forest MM9 1st Tra showed a composition of Proteobacteria (98.72%), Bacteriodetes (0.66%), Firmicutes (0.20%), Planctomycetes (0.02%), and Actinobacteria (0.07%). Populations of Germmatimonadetes, Chlorobi, WPS-2 and BRC1 were undetectable when the rhizosphere was cultured in the designed media. Likewise, the population of Verrucomicrobia, Acidobacteria, FBP, Armatimonadetes, OD1, Germmatimonadetes, TM6, Chlamyadiae, Chloroflexi, Chlorobi, WPS-2, and BRC1 was not detected either upon subsequent transfer forest to the forest MM9 1st Tra.
Population dynamics of upland and forest rhizosphere MF cultured in MM9 media at family level
The rhizosphere of the upland Hawaii 7996 plants harbored a total of 77 families. The cultures from upland MM9 maintained 39 (50.64%) from the original 77 families. Of the 39 families in upland MM9, the upland MM9 1st Tra maintained a total of 24 families (61.53% of MM9 culture and 31.1% of original population) (Supplementary Fig. 2 and Supplementary Table 4).
The rhizosphere of the forest Hawaii 7996 plants harbored a total of 82 families. The cultures from forest MM9 maintained 43 (52.4%) from the original 82 families. Of the 43 families in upland MM9, the forest MM9 1st Tra maintained a total of 23 families (53.4% of the MM9 culture and 28.04% of original population) (Supplementary Fig. 2 and Supplementary Table 4).
Top ten families across upland MM9 1st Tra and forest MM9 1st Tra
The top ten families among the upland MM9 1st Tra in a descending order are; Enterobacteriaceae (94.6%), Caulobacteraceae (1.29%), Brucellaceae (0.77%), Chitinophagaceae (0.68%), Sphingomonadaceae (0.39%), Bacillaceae (0.34%), Pseudomonadaceae (0.23%), Comamonadaceae hajeo(0.19%), Methylobacteriaceae (0.17%), and for forest MM9 1st Tra Enterobacteriaceae (78.96%), Pseudomonadaceae (18.37%), Sphingomonadaceae (0.66%), Sphingobacteriaceae (0.65%), Burkholderiaceae (0.34%), Paenibacillaceae (0.15%), Comamonadaceae (0.10%), Oxalobacteraceae (0.09%), Microbacteriaceae (0.07%), and Methylobacteriaceae (0.06%). Although the population of Enterobacteriaceae was highest in both the upland MM9 1st Tra and forest MM9 1st Tra, there was a significant difference between the population of the two communities (P = 0.007937) shown in Supplementary Fig. 1.
Shift in population reveals candidate family potentially responsible for the observed plant phenotype
Since the upland MM9 1st Tra showed maximum capacity to suppress the disease progress (Fig. 1) which instigated a further scrutiny into both; the structure of the community harvested from upland MM9 1st Tra and, its compositional differences from other treatments. The most evident difference in terms of the population composition observed at the family level (Supplementary Fig. 2 and Supplementary Table 4) was the population of Enterobacteriaceae (Supplementary Fig. 3). This allowed the inference of assumption to consider that Enterobacteriaceae may contribute to resistance against the pathogen. This further was supported by the disease progress curve (Fig. 1) which showed that the ability, to suppress disease, of bacterial community harvested from upland MM9 is less than that harvested from upland MM9 1st Tra but higher than forest MM9 and forest MM9 1st Tra.
Identification of OTU0 and its antagonistic activity against R. solanacearum
To isolate the bacterial strain representing OTU0, the higher dilutions (10−5-10−8) were plated for purpose of dilution to extinction of the low abundant bacterial strains, since the population of Enterobacteriaceae represented 94.6% of the total population. The serial dilutions were spread on five different media types; TSA, R2A, Marine agar, LB and NB agar. From each media, pure colonies were used for performing colony PCR amplification. The sequence from the colonies was identified as Enterobacter tabaci using Eztaxon database with 97% identity. Therefore, we tentatively designated this isolate as Enterobacter sp.
The activity of Enterobacter sp. against bacterial wilt suppression was assessed using three weeks Hawaii 7996. The disease progress for plants in non-sterilized soil treated with Enterobacter showed that this isolate confers disease suppression. Significant difference between disease index of treated and control plants was observed for bacterial wilt disease progress (P = 0.019471) (Fig. 5A). The activity of Enterobacter sp. against bacterial wilt suppression was assessed using three weeks old Hawaii 7996 in sterilized soil. The disease progress for plants treated with the Enterobacter planted in sterilized soil showed that the isolate did not confer disease suppression. No significant difference between disease index of treated and control plants was observed for bacterial wilt disease progress (P = 0.9615) (Fig. 5B). The activity of Enterobacter isolate against R. solanacearum in vitro was also assessed by examining by the zone of inhibition on agar plates. The Enterobacter isolate was seen to inhibit the growth of R. solanacearum which may be due to its ability to occupy niche which may then not be available to R. solanacearum, or has antagonistic activity (Supplementary Fig. 4).

Disease progress of R. solanacearum strains SL341 on tomato in non-sterilized (A) and sterilized (B) nursery soil. Thirty 21-days-old tomato plants (cv. Hawaii 7996) treated with bacterial cells of Enterobacteriaceae strain, control plants treated with 2.5 mM MES (pH 5.7) were inoculated by R. solanacearum SL341 at a final density of 1 × 107 cfu/g soil. Plants were rated for disease index through 14 days, vertical bars represent the standard error of the mean (n = 30). Significant difference was observed by repeated measures ANOVA between control and plants treated with Enterobacteriaceae strain (P = 0.019471) in non-sterilized soil. No significant difference was observed by repeated measures ANOVA between control and plants treated with Enterobacteriaceae strain (P = 0.9615) in sterilized soil.
Discussion
Previous studies employing the gnotobiotic plants with synthetic communities proposed that the establishment of simpler synthetic community is plausible to comprehend microbiota functions of the host-associated communities (Bai et al., 2015; Bodenhausen et al., 2014; Lebeis et al., 2015). Our approach in this study attempted to harvest the microbial community from tomato rhizosphere to have a relatively simpler bacterial community using mixed culture. We have investigated if the simpler cultured bacterial community still maintains the original microbiota function of the community to suppress the bacterial wilt disease. This is the first report to partially harvest the rhizosphere associated microbiota capable of retaining the bacterial wilt suppression function of the original microbiota of tomato rhizosphere.
Since, the microbial communities in the rhizosphere differ and depend on the resources, space and time (Fierer, 2007), MM9 media-based harvests was an idealistic approach to mimic such an environment and to allow culturing of maximum number of microorganisms from rhizosphere. The resultant communities harvested from the designed media were less diverse and simpler than those of the original MF. The discrepancy between the cultured bacterial population and the rhizosphere bacterial population is inevitable; the inability to culture the whole community has already been widely recognized (Hirsch and Mauchline, 2012). While an estimated less than 1% of the microorganisms can be grown routinely from various microbial ecosystem on the standard culture media (Amann et al., 1995), it has been also shown that over 66% plant associated microorganism can be cultured (Bai et al., 2015). However, previous approaches are usually dependent on agar medium to have pure cultures of single isolates. Our designed media however, shows an ability to support growth of microorganisms to a greater extent using mixed culture approach (Supplementary Table 4 and 5). The composition of the upland MM9 bacterial community harvested harbored 50.64% of the original upland rhizosphere community. The bacterial wilt incidence (Fig. 1) shows that there is significant difference between the occurrence of disease between control plants and plants under upland MM9 treatment and between control plants and plants under upland MM9 1st Tra treatment, but no significant difference between plants under upland MM9 treatment and plants under upland MM9 1st Tra treatment. This suggests that the community cultured from upland MM9 is capable of conferring the original microbiota function, i.e. bacterial wilt suppression. Not only has it the potential to confer bacterial wilt suppression but also has a larger bacterial community compared to the upland MM9 1st Tra community. Hence, this may be used as potential source of making a representative synthetic community.
A gargantuan number of microorganisms are associated with the plants, and to pin the properties of such humungous microbial community to specific roles is rather difficult (Berendsen et al., 2012). The relatively simpler community structures of upland and forest of MM9 media and subsequent cultures, gives an insight into the potential role of Enterobacteriaceae members possibly involved in suppressing bacterial wilt disease (Supplementary Fig. 2). Here, we isolated and identified a bacterial isolate that were predominant in the mixed culture. The 97% identity of 16S rRNA gene between the Enterobacteriaceae isolate and OTU0 sequence is due to the fact that the sequence for OTU0 is a representative sequence generated by the USEARCH algorithm. This step clusters the amplicon sequences and each cluster are defined by a centroid, which is the representative amplicon sequence and every sequence in the cluster has a similarity above the 97% identity threshold (Edgar, 2010). The treatment of Enterobacteriaceae isolate in non-sterilized soil successfully suppressed the occurrence of the bacterial wilt disease; however, its treatment in sterilized soil failed to confer the disease suppression (Fig. 5B). The phenomena can be attributed to the potential disease suppression ability of this strain in presence of other bacterial members as a community rather than independently. A member of family Enterobacteriaceae, Enterobacter cloacae, has been shown to act as keystone species in successfully inhibiting the disease in a model system of synthetic bacterial community (Niu et al., 2017). The results of this study provide a precedence to take into account the potential role of members of Enterobacteriaceae as keystone species which may help structure a community which is beneficial to the plants. The structure of the communities harvested from the MM9 1st Tra media for upland and those of forest differed. A list of the top ten families from both culture medium shows different families. Enterobacteriaceae, Pseudomonadaceae, Sphingomonadaceae and Comamonadaceae in both cultures represented the top ten families, but with different relative abundance values. The RA of most abundant family, Enterobacteriaceae, across both cultures was significantly different (Supplementary Fig. 3). The population of the common families varied between the cultured members from MM9 1st Tra for upland and those of forest MM9 1st Tra, and the members of both the communities were also different. The members of Proteobacteria, such as Pseudomonas and Enterobacter (Lwin and Ranamukhaarachchi, 2006) and Firmicutes, such as Bacillus (Chen et al., 2012; Da Silveira et al., 1995; Wydra and Semrau, 2005) and Pseudomonas spp (Lemessa and Zeller, 2007) has been reported as biocontrol agents against R. solanacearum. Although we have not isolated those bacteria from our mixed culture, such members seem to be also major bacterial members present in our MM9 mixed culture from the upland tomato rhizosphere.
In conclusion, MM9 medium approach was successful to cultivate a portion of the original microbiota of tomato rhizosphere, capable of retaining the bacterial wilt suppression trait. This approach can potentially be utilized to isolate potential key player of microbial community function for the construction of simpler synthetic bacterial communities. Yet, it remains to be evaluated what degree of synthetic microbial community harbored from the designed growth media is maintained in terms of community structure and the associated phenotypic traits in the plant rhizosphere.
Supplemental Materials
Acknowledgments
This research was supported by the Cooperative Research Program for Agricultural Science & Technology Development to S.-W. L. (PJ01093901) through Rural Development Administration, Republic of Korea.