Cucumber mosaic virus (CMV) is a type member of the genus Cucumovirus and has a very broad host range (Palukaitis and García-Arenal, 2003). Its genome comprises three single-stranded plus-sense RNAs, named RNA 1-3 in order of decreasing size, which encode five viral proteins: 1a, 2a, 2b, 3a (movement protein; MP), and capsid protein (CP). Initially, CMV strains were classified into two main subgroups, I and II, based on converging biological, serological, physical criteria (Hu et al., 1995; Ilardi et al., 1995; Wahyuni et al., 1992), nucleic acid hybridization (Owen and Palukaitis, 1988), and gene sequences (Owen et al., 1990; Sala and Bala, 1999). CMV strains were further divided into three subgroups including IA, IB, and II based upon phylogenetic analyses of a large number of the sequences of the CP gene as well as rearrangements in the 5′ non-translated region (NTR) of RNA3 (Roossinck et al., 1999). However, additional analyses with full-genome sequences of CMV strains demonstrated that some strains were grouped in neither subgroup I nor subgroup II but in a new subgroup, named subgroup III (Jacquemond, 2012; Liu et al., 2009); while some strains were in between subgroups I and II (Chen et al., 2007; Jacquemond, 2012; Maoka et al., 2010), and the others were in an intermediate position between subgroups IA and IB (Jacquemond, 2012; Lin et al., 2004; Masuta et al., 2002). Thus, analysis of at least one part of each genomic segment was necessary for confident genotyping of CMV strains (Jacquemond, 2012).
Ever since the first report of CMV strain isolated from soybean (Koshimizu and Izuka, 1958), several additional CMV strains have been isolated (Hong et al., 2003; Senda et al., 2004; Takahashi et al., 1980). The firstly isolated CMV soybean strain induced mild mosaic and stunting on infected soybean plants and was transmissible through seeds with a high efficiency by aphids (Koshimizu and Iizuka, 1963; Takahashi et al., 1980). Even though the biological characteristics of CMV soybean strain have been studied extensively, there was no available sequence information on CMV soybean strain genomes. A previous report analyzing the phylogenetic relationship of the nucleotide sequences of the 3a and CP genes of seven CMV soybean strains showed that these CMV soybean strains formed a distinct cluster separated from the other CMV strains (Hong et al., 2003).
Construction of infectious cDNA clones corresponding to the genomes of RNA viruses is necessary since viral cDNA clones are essential sources for maintaining their original RNA genomes and for engineering viral vectors (Boyer and Haenni, 1994; Nagyová and Subr, 2007). In addition, easy manipulation of virus cDNA clones allows mutagenesis (deletions, insertions, substitutions) of specific regions of virus genomes and subsequent introduction of the mutated or wild type genomes into host plants for studies of virus life cycle, functions of viral proteins and their interactions with various host factors involved in virus replication, cell to cell movement, resistance responses (Bendahmane et al., 2002; Kim and Palukaitis, 1997; Paalme et al., 2004; Prüfer et al., 1995).
In this study, we obtained a CMV isolate from Glycine soja (wild soybean) and named CMV-209. Using this isolate, we generated infectious full-genome cDNA clones of CMV-209. Using the cloned CMV-209, its molecular characteristics were examined by determining complete nucleotide sequences of each genomic RNA segment and comparing to those of the other representative CMV strains. The biological characteristics were investigated by inoculating the cloned CMV-209 in various legume species as well as Nicotiana benthamiana.
Materials and Methods
Virus source and construction of the full-length cDNA clones of CMV-209
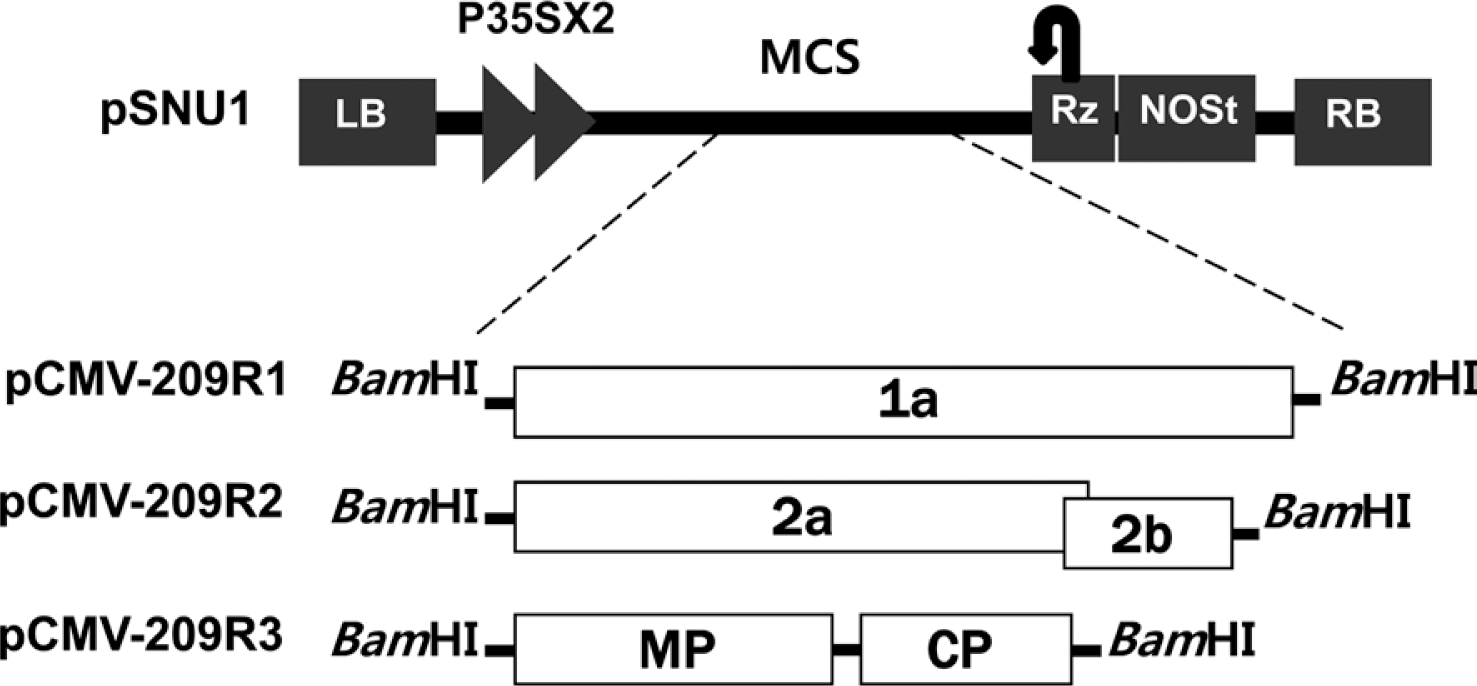
Wild soybean samples showing typical mosaic symptoms were collected in Suwon, Korea 2006. To find the samples infected with CMV, the collected samples were screened by RT-PCR using the CMV specific primers CMV R3 561 Fw and CMV R3 1473 Rv (Table 1). One (sample No. 209) of the wild soybean samples was found to be infected with CMV. Total RNA was extracted from the CMV-infected wild soybean (No. 209) using TRI Reagent method (MRC, USA) and subjected to RT-PCR to amplify full-length cDNAs corresponding to CMV RNA1, RNA2, and RNA3 using the specific primer pairs that contain BamHI restriction site (Table 1). The amplicons were digested with BamHI and ligated with the modified binary vector pSNU1 (Park and Kim, 2006) which was digested with BamHI. The resulting clones carrying the full-length cDNA of RNA1, RNA2 or RNA3 of CMV were named as pCMV-209R1, pCMV-209R2 and pCMV-209R3, respectively (Fig. 1).
Infectivity test and screening host range for CMV-209
All of the CMV constructs (i.e. pCMV-209R1, pCMV-209R2, and pCMV-209R3) were individually transformed into Agrobacterium tumefaciens strain GV2260 by electroporation and cultured as previously described (Park and Kim, 2006). The Agrobacterium transformants were grown at 30°C until OD600 = 0.5 ± 0.05. A mixture of three Agrobacterium cultures harboring constructs pCMV-209R1, pCMV-209R2, and pCMV-209R3 in a 1:1:1ratio to yield a total OD600 of 0.17 ± 0.02 was used for co-agroinfiltration. This final Agrobacterium mixture was infiltrated into the abaxial surface of the N. benthaminana leaves (3 to 4 weeks old) by using a 1-ml syringe without a needle. pCMV-Fny clones (pCR1(+), pCR2(+), and pCR3(+); Seo at al., 2009) were also included in agroinfiltration as control. After 7 days, the upper noninoculated N. benthamiana leaves were subjected to RT-PCR using the primer pairs CMV detR2 F and CMV detR2 R (Table 1) to examine whether the infiltrated plants are systemically infected with CMV-209. As shown in Table 2, the sap extracts from the N. benthamiana plants systemically infected with CMV-209 were used as viral inoculums for sap-inoculation of 14 legume cultivars including Pisum sativum (pea), Vigna sinensis (cowpea), V. angularis (cv. Chungbupat), Phaseolus vulgaris (Kidney bean), Ph. vulgaris (Pinto bean), Glycine max (cv. Somyong), G. max (cv. Pureunkong), G. max (cv. Sowon), G. max (cv. Danbaek), G. max (cv. L29), G. max (cv. Daewon), G. max (cv. V94-5152), G. max (cv. Huanggumkong), G. max (cv. Lee74), and G. soja (wild soybean).
Sequencing, comparative, and phylogenetic analyses
The full-length cDNA sequences in the infectious clones of CMV-209 (pCMV-209R1, pCMV-209R2 and pCMV-209R3) were sequenced using the appropriate primers (the list of primers used for the sequencing is available on request). The nucleotide sequences and deduced amino acids sequences of CMV-209 were compared with those of other representative CMV strains retrieved from the GenBank database. Sequences of PSV-P, PSV-W and PSV-Mi as well as TAV-KC were used as out-groups (Table 3). Multiple sequence alignments of all the analyzed sequences were obtained using CLUSTALS (Thompson et al., 1997). The sequence similarity comparisons were performed using Lasergen Package. The phylogenetic analyses were carried out using the Neighbor-Joining method (Saitou and Nei, 1987). The confidence probability was estimated using the bootstrap test (1000 repetitions) (Dopazo, 1994; Felsenstein, 1985; Rzhetsky and Nei, 1992). The evolutionary distances were computed using the p-distance method (Nei and Kumar, 2000) and were the units of the number of nucleotide or amino acid differences per site. Evolutionary analyses were conducted in MEGA6 (Tamura et al., 2013).
Results and Discussion
Infectivity and host range of CMV-209
To verify the infectivity of the full-genome cDNA clone of CMV-209, we first infiltrated N. benthamiana plants with a mixture of agrobacterium cultures harboring pCMV-209R1, pCMV-209R2, and pCMV-209R3. All the infiltrated plants showed very mild or no visible symptom (Fig. 2B) while showing typical symptoms including mosaic, stunting, or distortion of the leaves from CMV-Fny infiltrated plants (Fig. 2C). Although there was no obvious visible symptom from CMV-209 infiltrated plants, progeny viruses were detected from the upper systemic leaves by RT-PCR using CMV-specific primers (Fig. 2D) demonstrating that the full-genome cDNA clones of CMV-209 are fully infectious.
Next, since CMV-209 was isolated from wild soybean, the host range and infectivity of CMV-209 were examined in various legume species. To this end, twelve legume species were inoculated with the sap extracted from the N. benthamiana plants systemically infected by the CMV-209 infectious clone. Among the tested legume species, only G. soja (wild soybean) and P. sativum (pea) were systemically infected with CMV-209 (Table 2). The pea plants infected with CMV-209 showed severe stunting symptom (Table 2; Fig. 3A), rosetting of stem (Fig. 3B) and their roots were significantly shorter and fewer than those of healthy and CMV-Fny infected pea plants (Fig. 3A). CMV-209 inoculated Vigna sinensis (cowpea) showed necrotic spots on inoculated leaves (Fig. 3C).
Host adaptation is one of the main factors for sequence convergence or divergence of viruses. CMV-209 could systemically infect pea (P. sativum) and N. benthamiana, but not the other host plants tested in this study, suggesting that CMV-209 is host-selective strain (Table 2). CMV-209 seems to have adapted itself to infect pea because other typical CMV strain such as CMV-Fny could not infect pea (Fig. 3A). In fact, Hong et al. (2003) also reported that CMV soybean strains might have adapted to soybeans in their evolutionary history and that they had evolved faster than CMV subgroup IA.
Interestingly, CMV-209 also induced necrotic spots in inoculated leaves of V. sinensis as other CMV strains did (Fig. 3C). Although two amino acid residues of the 2a protein at positions 631 and 641 being Phe and Ala, respectively, were also conserved in CMV-209, which have been reported to be crucial for causing a local hypersensitive response in cowpea (Kim and Palukaitis, 1997), it remains to be determined whether these two conserved amino acid residues are actually responsible for causing necrotic local lesions. The difference of host range and/or infectivity between the CMV-209 and other reported CMV strains in tested host plants observed in this study might be due to sequence changes in CMV-209 RNA genome segments. In this regard, it is worth mentioning that the RNA3 segment including 3a and CP proteins has been reported as a host range determinant and thus might be more responsible as host-interactive constraints than the other viral gene products (Hong et al., 2007; Hong et al., 2003; Roossinck, 2002; Ryu et al., 1998). We are currently conducting experiments with reasserted RNA segment using several CMV strains including Fny and 209 to further determine whether RNA3 segment is responsible for determining host range and/or infectivity.
Molecular characteristics of CMV-209
The complete nucleotide (nt) sequence of CMV-209 was determined and deposited in the GeneBank database (Accession Nos: KJ400002, KJ400003, KJ400004). The sizes of the genomic RNAs varied for the different strains and species. The sizes of RNAs 1, 2, and 3 of the strains in CMV subgroup I were in range of 3357-3365 nt, 3036-3060, and 2213-2220 nt, while in CMV subgroup II were 3389-3391, 3038-3053, 2197-2209 nt, respectively (Palukaitis and García-Arenal, 2003). The length of RNA1, RNA2, and RNA3 of CMV-209 were 3,360 nt, 3,057 nt, and 2,243 nt, respectively, indicating that CMV-209 belongs to CMV subgroup I.
The coding part of the genome consists of five ORFs, i.e. 1a, 2a, 2b, MP, and CP. The first ORF encodes the largest protein (111.65 kDa; 993 aa) which possesses a putative methyltransferase domain and a helicase motif. The second ORF codes the RNA-dependent RNA polymerase (RdRp) protein (97.043 kDa; 858 aa). The third ORF encodes the smallest protein (12.846 kDa; 110 aa), which has function as a silencing suppressor. The fourth is a putative MP (30.595 kDa; 279 aa) and the fifth is the 24.136 kDa CP consisting of 218 aa.
The nucleotide positions of the five ORFs (1a, 2a, 2b, 3a, and CP) for CMV-209 and length of which were presented in Table 4. The initiation codon for the 1a, 2a, 2b, 3b, and CP genes of CMV-209 were positioned at position 95, 78, 2413, 110, and 1280 compared with 95-98, 85-87, 2417-2419, 120-121, and 1257-1260 of subgroup IA and 95-96, 79-80, 2414-2415, 122, and 1255-1261 of subgroup IB, respectively. The size (2577 nt) of CMV-209 ORF2a was slightly different from subgroups IA and IB, which was 2574 nt in length. The length of CMV-209 ORF 3a (840 nt) were same as subgroup IA (840 nt) but slightly differed from that of subgroup IB (852 nt). ORFs 1a, 2b, and CP of CMV-209 were 2982 nt, 333 nt, 657 nt, respectively, in length as were in subgroups IA and IB (data not shown). Further information of the other components of RNAs was indicated in detail in Table 4.
Comparative analysis of the CMV-209 genome sequence
According to the International Committee on Virus taxonomy (ICTV) criteria for discriminating species from the genus Cucumovirus, subgroups ordinarily have at least 65% sequence identicalness. The nt sequence similarity for the complete genomic RNAs among the CMV-209 and the representative CMV strains was ranged from 71.8 % to 93.4% while the similarity among CMV-209 and other cucumoviruses such as PSV and TAV was in the range of 56.1-68.7% (Table 5). The genome sequence of CMV-209 had highest similarity with that of CMV-Fny (Table 5).
In CMV, within-subgroup nt homologies of subgroup II and III were very high (more than 98% and 94%, respectively). The nt sequence of CMV-209 had the sequence identity to subgroup II and III only around 76% and 89.5%, respectively (data not shown). In the case of subgroup I, the sequence identities of RNA1, 2, and 3 within subgroup IA were more than 92.7, 95.8, and 97.1%, respectively, and were more than 97.8, 91.2 and 97.4%, respectively, within subgroup IB. The sequence identities of RNA1, 2, and 3 between sister group, subgroup IA and IB, were in the range of 89.8-92.2, 89.1-91.5, and 91.8-93.9%, respectively (data not shown).
Genomic segment comparison of CMV-209 with the strains in subgroup I, including subgroup IA and IB, showed their identity levels of 89.6-91.2, 90.0-92.4, and 91.7-93.4% in RNA1, 2, and 3 sequences, respectively (Table 5). The 3′UTR of CMV has information required for minus strand synthesis of the viral RNA during replication and is highly conserved within subgroup. However, the 3′UTR nt sequences of CMV-209RNA1, RNA2, and RNA3 showed the similarities of 87.5-91.1, 86.0-88.7, and 92.0-93.7%, respectively, when compared to those of subgroup IA and IB (Table 5). Taken together, our analyses suggested that CMV-209 could belong to a distinct sister group from subgroup IA and IB.
Phylogenetic analysis
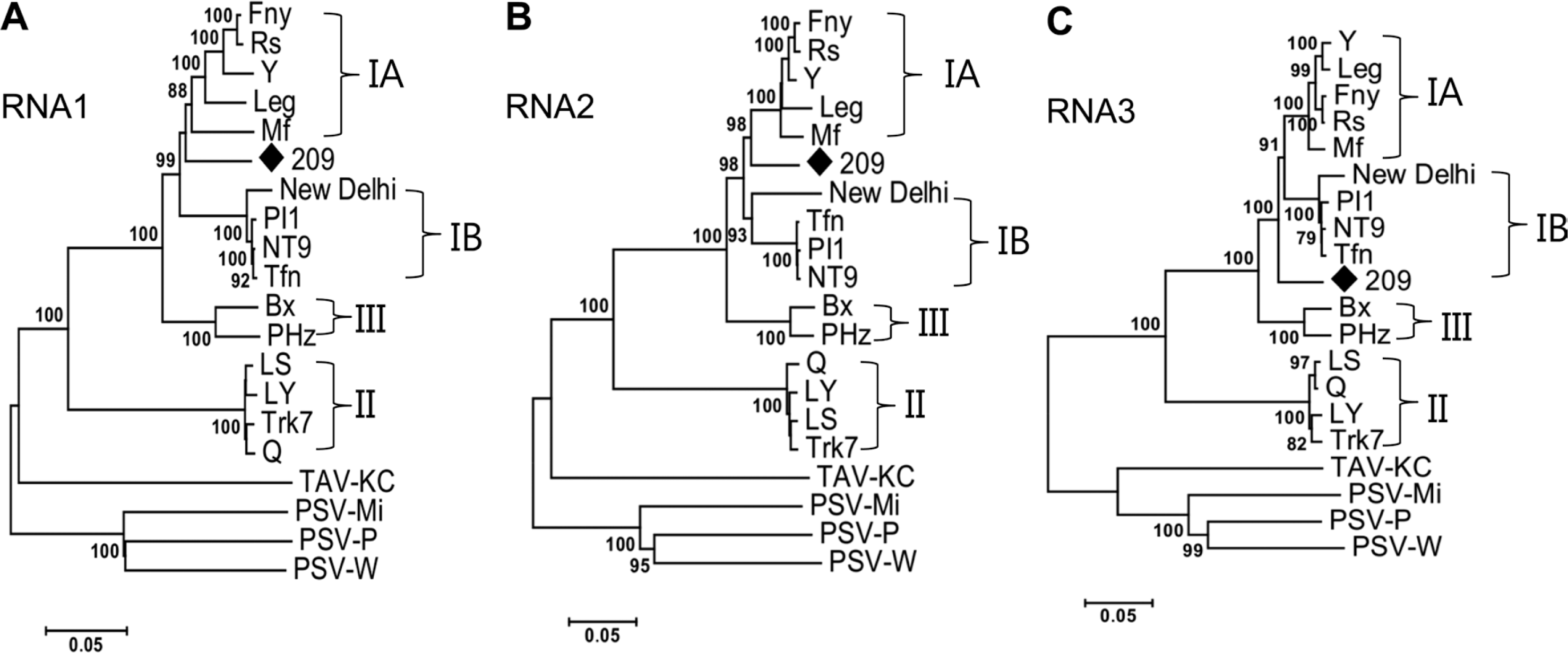
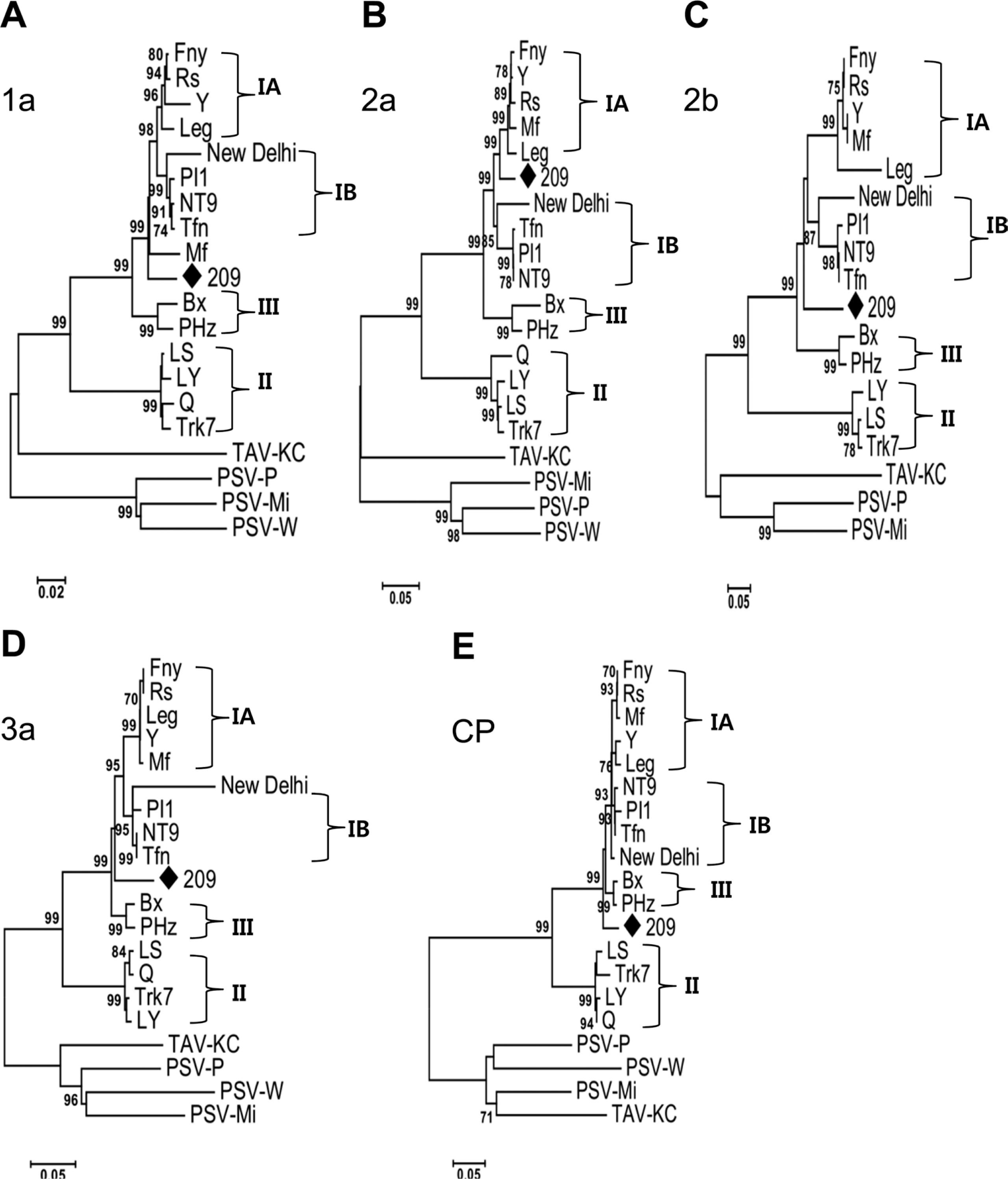
First, phylogenetic trees for each CMV-209 genomic segment were constructed, in which PSV and TAV were used as out groups. The trees revealed obviously that CMV-209 was clustered in subgroup I. For all three RNAs, a first branching distinguished from clear clustering of group II strains, a second one separated into subgroup III, and the next branching of the trees was precisely divided into subgroups IA, IB, and CMV-209. In the case of RNAs 1 and 2, CMV-209 shared same branching points to subgroup IA (Fig. 4A, B), suggesting that these two RNAs of CMV-209 had closer relationship to subgroup IA than subgroup IB. Although RNA3 of CMV-209 had the same progenitor with subgroup IA and IB, it formed a distinct branch before separation of branches of subgroup IA and IB (Fig. 4C).
Next, phylogenetic analyses of 1a, 2a, 2b, 3a, and CP genes based on amino acid sequences were established. The 2a and 3a protein trees had identical patterns with the full length RNA 2 and 3 nt sequence trees, respectively (Fig. 5B, D). The 1a and 2b protein trees presented a complete segregation of CMV-209 out of subgroups IA and IB (Fig. 5A, C), whereas 1a and 2b ORF nt sequence trees (data not shown) and genomic RNA 1 and 2 trees showed CMV-209 close to subgroup IA. Both CP aa sequence tree and CP nt sequence tree (data not shown) exhibited that CMV-209 was located in a new branch separated from subgroups IA and IB; however, CP protein tree gave a clearer separation. This may suggest that the difference of the biological characteristics among CMV-209 and subgroups IA and IB appeared at amino acid level. In general, amino acid phylogenetic analyses of CMV-209 and the CMV strains had unquestionably placed CMV-209 in another subgroup of subgroup I.
In summary, our analyses provide molecular evidences that CMV-209 may belongs to a distinct subgroup within CMV subgroup I and that the adaptation of CMV-209 in pea (P. sativum) could be due to aa changes of RNA 3 segment based upon sequence analyses. Further study will test whether RNA3 is indeed responsible for adapting CMV-209 in pea by inoculating swapped RNA segments with the other CMV strains and mutant clones which contain site-directed mutations.


 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print