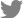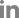Acidovorax citrulli (Ac) is a Gram-negative, plant pathogenic bacterium that causes bacterial fruit blotch (BFB) on cucurbit crops, leading to serious crop losses and a subsequent damage to the cucurbit industry (Bahar and Burdman, 2010). Since Ac is a seed-borne pathogen, the disease cycle of BFB begins with infected seeds (Song et al., 2020). It is mainly spread by rain splash; the dispersed Ac can enter plants through open stomata or through stigmas, thereby leading to a disease outbreak (Johnson et al., 2011). The symptoms of BFB include a water-soaked lesion on the cotyledons and wilting in the initial stage (Burdman and Walcott, 2012). Furthermore, in the infected fruits, water-soaked lesions appear on the fruit surface, finally leading to necrosis of the internal fruit parts. However, in spite of the importance of developing cultivars resistant to Ac infection, it has not yet been identified.
Several virulence factors of Ac and their related mechanisms have been characterized in previous studies. Similar to other plant pathogenic bacteria, the type II and III secretion systems of Ac are crucial for its virulence (Johnson et al., 2015; Zhang et al., 2018). Moreover, the type IV pili-mediated motility and quorum-sensing are required for its virulence and seed-transmission (Bahar et al., 2009; Johnson and Walcott, 2013). Recently, two proteins, bifunctional chorismate mutase/prephenate dehydratase (CmpAc) and glycerol-3-phosphate dehydrogenase (GlpdAc), have been identified in Ac (Kim et al., 2020b, 2021). Both proteins are involved in its virulence as well as in other mechanisms, such as twitching motility, tolerance against external stresses, or growth in diverse carbon sources. This suggests that CmpAc and GlpdAc display pleiotropic effects in Ac. However, other virulence factors of Ac and their related mechanisms are poorly understood.
Aminotransferases form a group of enzymes that can remove the amino group from amino acids, transfer it to α-keto acids, and thereby produce new amino acid products (Shin et al., 2018). Specific aminotransferases have been identified for all amino acids, except lysine, threonine, proline, and hydroxyproline (Ndrepepa and Kastrati, 2019). Apart from regulating amino acid metabolism, aminotransferases are mainly involved in the tricarboxylic acid (TCA) cycle. Some aminotransferases, namely the pyridoxal phosphate-dependent aminotransferases (Ppda), require pyridoxal phosphates as coenzymes for their enzymatic activity (Hafkenscheid and Dijt, 1979). In Mycobacterium tuberculosis, aminotransferases are required for growth and virulence in mice (Jansen et al., 2020). Moreover, the aminotransferases of Xanthomonas oryzae pv. oryzicola, which causes the bacterial leaf streak disease in rice, are also involved in virulence (Guo et al., 2012). However, the role of Ppda in Ac has not yet been reported.
We performed two different virulence tests to determine the involvement of pyridoxal phosphate-dependent aminotransferase in Ac (PpdaAc) in its virulence. Additionally, the biological and cellular mechanisms related to PpdaAc were predicted using comparative proteomic analysis and clusters of orthologous groups (COG) classification. To identify the genes that are associated with virulence in Ac, we screened the Tn5-insertional mutant library, as described previously (Kim et al., 2020b). We identified a virulence-deficient mutant in which a putative pyridoxal phosphate-dependent aminotransferase gene (ppdaAc) was disrupted by Tn5, according to the manufacturer’s protocol (Lucigen, Middleton, WI, USA), and we named it AcΔppdaAc. To generate the complemented strain, the open reading frame of ppdaAc was amplified using a specific primer set (5′-GGTACCATGAAGACGATCCACAAGTC-3′ and 5′-CGGGCGGCTGCCGAGCACCACCACCACCA CCACTGAGAGCTC-3′) and cloned into pGem-T easy vector (Promega, Madison, WI, USA); the gene sequence was confirmed using Sanger sequencing. The confirmed gene was cleaved by KpnI and SacI and ligated into the broad-host-range vector pBBR1-MCS5, thereby creating the recombinant construct pMCS5-ppdaAc. Thereafter, the construct was introduced into the wild-type Ac, generating AcΔppdaAc(PpdaAc). To minimize the side effects of vector insertion, an empty vector was introduced into the wild-type and AcΔppdaAc strains, creating Ac(EV) and AcΔppdaAc(EV), respectively. All the plasmids and bacterial strains used in this study are listed in Table 1.
To test the pathogenicity, we performed both germinated- seed inoculation and leaf infiltration, as described previously (Kim et al., 2020b). In case of germinated-seed inoculation, all the seedlings infected by Ac(EV) were completely wilted, whereas in case of AcΔppdaAc(EV) inoculation, the disease symptoms were absent (Fig. 1A and B). The virulence of the complemented strain was restored to levels similar to that of Ac(EV). At 7 days post inoculation, the disease indexes of Ac(EV), AcΔppdaAc(EV), and AcΔppdaAc(PpdaAc) reached 2, 0, and 1.6, respectively. Incidentally, the results obtained from the leaf infiltration experiment were similar to those obtained from the germinated- seed inoculation (Fig. 1C and D). The population of AcΔppdaAc(EV) was significantly lower than those of the others in the entire period. Leaves infiltrated by Ac(EV) and AcΔppdaAc(PpdaAc) exhibited dark colored blight symptoms, while those infiltrated by AcΔppdaAc(EV) remained green. Therefore, it seemed that the virulence of AcΔppdaAc(PpdaAc) had been partially complemented. This partial complementation may be attributed to the different promoters and/or copy numbers of the gene in AcΔppdaAc(PpdaAc) because pBRR1-MCS5 maintains several copy numbers in bacteria, and the expression of the targeted gene is driven by the lacZ promoter, which is constitutively expressed (Kovach et al., 1995). Hence, these data demonstrate that PpdaAc is indispensable for virulence in Ac, which is in accordance with previous studies that have established the involvement of Ppda in virulence of other pathogens. For instance, in the Ppda deletion mutant of Ralstonia solanacearum the virulence of the pathogen is attenuated (Kai et al., 2016). Similarly, in Streptococcus pneumoniae, Ppda is important for the biosynthesis of cell wall (Han et al., 2018), thereby indicating that Ppda is one of its virulence factors.
It is well-known that one protein is involved in virulence and other distinct mechanisms in animal and plant pathogenic bacteria (Felgner et al., 2016; Kim et al., 2020b; Park et al., 2021). Therefore, we aimed to determine PpdaAc-related mechanisms by comparing the abundance of different proteins in Ac and AcΔppdaAc strains. Sample preparation, protein extraction, peptide generation, liquid chromatography with tandem mass spectrometry, and protein identification/quantification were performed based upon previously established protocols (Kim et al., 2020b). A total of 896 and 947 proteins were commonly detected in the three biological replicates of the wild-type and the mutant, respectively (Supplementary Table 1). Subsequently, these proteins were subjected to a comparative analysis, the results of which showed that 66 and 115 proteins were exclusively present in Ac and AcΔppdaAc, respectively; on the contrary, with respect to shared proteins, 35 and 68 proteins were more abundant (>2-fold) in Ac and AcΔppdaAc, respectively (Fig. 2A). Incidentally, PpdaAc (ATG95109) was detected only in the wild-type and not in the mutant, thereby indicating that the proteomic analysis had been performed successfully. Thereafter, these differentially abundant proteins were classified (Fig. 2B, Supplementary Tables 2 and 3) using the COG categorization (Tatusov et al., 2000). The proteins belonging to group C (energy production/conversion), E (amino acid transport/ metabolism), G (carbohydrate transport/metabolism), H (coenzyme transport/metabolism), I (lipid transport/metabolism), N (cell motility), Q (secondary metabolite bio-synthesis/transport/catabolism), and T (signal transduction) were highly abundant in the mutant, whereas, the proteins categorized in group F (nucleotide transport/metabolism), J (translation), L (DNA replication/recombination/repair), O (post-translational modification, protein turnover, and chaperones), and P (inorganic ion transport/metabolism) were more abundant in the wild-type. The representative proteins and their respective predicted mechanisms, based upon the proteomic analysis, are described in Fig. 3.
Living organisms acquire energy through diverse energy production pathways, including glycolysis (group G), TCA cycle (group E), and electron transport system (group C) (Fernie et al., 2004). The results of the proteomic analysis showed that the mutant strain expressed the proteins included in these groups more abundantly, as compared to the respective protein expressions in the wild-type. Moreover, gluconeogenesis-related proteins, namely glucose-6-phosphate isomerase (ATG97016) and fructose 1, 6-bisphosphatase (ATG94560), were more abundant in the mutant than in the wild-type. Furthermore, isocitrate lyase (ATG96777) that is essential in the glyoxylate pathway, an alternative pathway for carbon metabolism (Chew et al., 2019), was more abundant in AcΔppdaAc than in the wild-type. Normally, isocitrate is utilized in the TCA cycle; however, when bacteria utilize fatty acids for growth, they use the isocitrate in the glyoxylate pathway for gluconeogenesis (Gonçalves et al., 2016). Since our results show a greater abundance of fatty acid-related proteins in the mutant than in the wild-type, it may be predicted that PpdaAc affects gluconeogenesis from fatty acids.
Previous studies have shown that Ppda is associated with glutamate production (Ndrepepa and Kastrati, 2019). In this study, we observed that glutamate 5-kinase (ATG93720), which is involved in glutamate synthesis, was more abundant in the wild-type than in the mutant. In biochemical pathways, glutamates are converted to aspartates, which, in turn, can be degraded to homoserine by homoserine dehydrogenase; the homoserines can be further converted to methionine and threonine (Kim et al., 2020a). Based upon our data, the enzymes necessary for the above-mentioned biochemical pathways, homoserine dehydrogenase (ATG95108), threonine synthase (ATG95107), and methionine synthase (ATG95998), are more abundant in the wild-type than in the mutant. Therefore, it can be postulated that glutamate synthesis would be significantly reduced in the mutant due to the lack of PpdaAc expression. This may lead to the increased expression of other enzymes responsible for glutamate production, such as glutamine amidotransferase, in the mutant. Incidentally, glutamine amidotransferase (ATG95676), which is involved in glutamate production, was more abundant in AcΔppdaAc than in the wild-type. Electron transfer system has four oxidoreductase complexes, namely NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), cytochrome c reductase (complex III), and cytochrome c oxidase (complex IV) (Schertl and Braun, 2014). In the proteomic analysis, we detected the presence of succinate dehydrogenase (ATG93227), NADH-quinone oxidoreductase subunit (ATG96994), flavoprotein subunit alpha (ATG93258), and glycerol-3-phosphate dehydrogenase/oxidase (ATG95651), which are associated with complex I and II (Fig. 3), and cytochrome c oxidase subunit II (ATG93913), cytochrome C4 (ATG93956), and 2Fe-2S ferredoxin (ATG95897), which are associated with complex III and IV. These results indicate that PpdaAc is related to amino acid metabolism as well as electron/energy production.
Since the abundance of the diverse proteins categorized in cell motility was altered (Fig. 2B), it is possible that PpdaAc is involved in bacterial motility; for example, flagella component proteins (ATG96515 and ATG96513) and pili-related proteins (ATG95316, ATG93727, ATG95626, and ATG95628) were more abundant in the mutant than in the wild-type (Supplementary Tables 2 and 3). Both flagella- and pili-dependent motilities are tightly associated with virulence in Gram-negative bacteria including Ac, Salmonella enterica, and Pseudomonas aeruginosa (Bahar et al., 2009; Giltner et al., 2010; Kim et al., 2020b; Yang et al., 2012). However, virulence decreases when a flagella-related gene is overexpressed (Yang et al., 2012). Chemotaxis-related proteins (ATG93311, ATG95368, ATG96536, ATG96859, ATG96534, ATG95814, ATG96440, ATG93889, and ATG96597) were also identified in the comparative proteomic analysis. Chemotaxis is the bacterial movement towards particular environmental cues using the motility functions (Wadhams and Armitage, 2004). The CheW is a crucial protein with respect to chemotactic movement in E. coli and Rhodobacter sphaeroides (Hamblin et al., 1997; Sanders et al., 1989). Based upon our results, the CheW (ATG96534) protein was more abundant in the wild-type. Hence, it may be inferred that PpdaAc affects the regulation of motility-related proteins, which, in turn, may contribute to virulence in Ac.
In the proteomic analysis, we detected the presence of proteins of the heat shock protein 70 family (ATG97035 and ATG93774) as well as molecular chaperones DnaJ (ATG95120) and HtpG (ATG96611) (Fig. 3). These proteins are associated with protection against stress conditions and virulence in pathogenic bacteria (Cui et al., 2017; De Maio, 1999; King et al., 2014). Additionally, several proteins related to inorganic ion transport/metabolism were also identified, especially diverse TonB-dependent receptors (ATG94456, ATG96986, and ATG97071) in the wild-type; these receptors are used for obtaining iron via siderophores (Fujita et al., 2019). In Acinetobacter baumannii and Pseudomonas fluorescens, mutation of TonB-dependent receptors triggers reduced biofilm formation, adherence, and virulence (Abdollahi et al., 2018; Hu et al., 2012). The biopolymer transporter ExbD (ATG94445) that can form a complex with TonB (TonB-ExbD complex) and transport the iron-siderophore into the cytoplasm through the cell membrane using proton motive forces (Ollis and Postle, 2012) was also detected. The exbD-knockout mutant of Pasteurella multocida shows reduced virulence (Bosch et al., 2002). However, a two-component system response regulator (ATG93349) was only identified in the comparative proteomic analysis, indicating that PpdaAc may not have a significant role in the signaling pathway. Therefore, our results suggest that PpdaAc has a role in post-translation modifications and ion transport but not in the signal transduction system.
In this study, we performed two different pathogenicity tests and demonstrated that PpdaAc is essential for the virulence of Ac. To determine the biological and cellular mechanisms associated with the PpdaAc protein, we compared the patterns of protein expression between the wild-type and the mutant using proteomic analysis. The comparative analysis revealed that PpdaAc is associated with virulence as well as other biological and cellular mechanisms, thereby suggesting that PpdaAc may have pleiotropic effects in Ac. Nevertheless, it is still unclear how the PpdaAc protein is involved in the predicted mechanisms. Hence, further studies need to be performed to elucidate the specific functions of PpdaAc at molecular and phenotypic levels. Those results will provide a new perspective of the mechanisms/ functions related to Ppda in Gram-negative bacteria.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print