Bacterial leaf blight caused by the plant pathogenic bacterium Xanthomonas hortorum pv. carotae (Xhc) was first described in carrots (Daucus carota L. subsp. sativus Hoffm.) in 1934 (Kendric, 1934). Bacterial blight caused by Xhc has become established globally, and is frequently observed in carrot crops in Europe, North America, and Asia. In Korea, carrot bacterial blight caused by Xhc has been designated and managed as a plant quarantine pathogen since 1996. The first Korean outbreak of Xhc was observed in a carrot field on Jeju Island in 2012, with a subsequent outbreak seen in two Jeju carrot fields in 2013 (du Toit et al., 2014; Myung et al. 2014; Pruvost et al., 2010).
Much of the global carrot seed supply is supplied by New York State in the United States. The US carrot seed supply produces 60% of seeds used to produce carrot root crops in Korea and up to 40% of the world’s supply of carrot seed. Harvested plant seeds can be a source of Xhc transmission, and infected seeds can act as a distribution mechanism by which Xhc can spread to new countries (du Toit et al., 2005). Symptoms of bacterial blight in carrot include small, irregular yellow lesions on the leaves, stems, and petioles that may appear as water-soaked necrotic lesions (Gilbertson, 2002). X. hortorum is one of several plant pathogenic Xanthomonas species and requires phylogenetic identification in addition to symptomatic characterization. Several studies have sequenced the 16S rRNA and 16S-23S internal transcribed spacer regions in the Xanthomonas genus (Adriko et al., 2014; Hauben et al., 1997; Maes, 1993), and phylogenetic classification of the Xanthomonas genus can also be achieved using multilocus sequence analysis (MLSA) (Parkinson et al., 2007; Young et al., 2008). X. hortorum was defined using DNA hybridization studies within the Xanthomonas genus (Vauterin et al., 1995). Subsequent phylogenetic and multi-position sequencing studies using gyrB or four housekeeping genes confirmed that X. hortorum grouped with X. cynarae and X. gardneri to form diverse clades (Young et al., 2008). However, more accurate chromosome mapping is needed for precise pathogen diagnosis and to facilitate Xhc characterization and evolutionary analysis. Currently, three Xhc assemblies are available in GenBank, one with a chromosome assembly and two with unassembled contigs (National Center for Biotechnology Information [NCBI] Xanthomonas hortorum pv. carotae database). However, a complete genome assembly with improved sequence quality and fewer contigs is essential to fully understand the species-specific Xhc genome and virulence diversity among highly conserved Xhc strains. In this study, we report a complete genome sequence of Xhc from Korea with high resolution. There have been no further reports of Xhc in Korea to date; however, to prepare for the risk of domestic outbreaks, a disease survey was conducted in 159 farms in eight cities and counties in the main carrot producing regions in 2020.
Xanthomonas strains were isolated from plants with bacterial leaf blight lesions from carrot farms in Korea (Fig. 1A). The sample surveyed a total of 159 farms in 8 cities and 5 provinces (Supplementary Table 1). Collected carrot leaves with bacterial blight symptoms were first surface-sterilized with 70% ethanol, and then disease-lesion border regions were excised, cut into small pieces, and immersed in 500 μl sterile water for 30 min. Bacteria were isolated by cultivating the immersion solution on yeast extract-dextrose-CaCO (YDC) medium agar at 27°C for 4 days.
Genomic DNA was extracted from isolated bacterial cultures using a Bacterial Genomic DNA Isolation kit (NORGEN Biotek, Thorold, Canada). Extracted DNA was polymerase chain reaction (PCR)-amplified using GoTaq Flexi4 DNA Polymerase (Promega, Madison, WI, USA). PCR amplification of Xanthomonas chaperone protein (dnaK), DNA gyrase subunit B (gryB), TonB dependent receptor (fyuA), and RNA polymerase sigma factor (rpoD) genes was performed as described previously (Young et al., 2008) (Supplementary Table 2). PCR conditions were as follows: 95°C for 5 min, 35 cycles of 95°C for 30 s, 60°C for 60 s, and 72°C for 1 min, followed by 72°C for 10 min. Sequences of the dnaK, gryB, fyuA, and ropD genes were collected from 30 Xanthomonas strains from NCBI and PAMDB (http://genome.ppws.vt.edu/cgi-bin/MLST/home.pl) and from Xhc strains isolated from Jeju Island in Korea. The four gene sequences were linked to create a base sequence of 4,620 bp and aligned using BioEdit 7.2.5. A phylogenetic tree was created for the aligned nucleotide sequences by repeating the bootstrap number of 1,000 using the maximum likelihood method using the MEGA 7 program. Isolated bacteria was found that exhibited high similarity to the previously sequenced Xhc M081 strain (GenBank accession no. AEEU01000001) (Fig. 1B).
For pathogenicity analysis, carrot seeds were planted in topsoil and cultivated at 25°C with 60% relative humidity until four or more true leaves had developed. Seedlings were then inoculated with 1 μl bacterial culture (O.D.600 = 0.5) and lightly wounded by stabbing the back of the leaf with a sterile 1 ml injection needle. Inoculated plants were incubated in a transparent plastic box for 2 days, and then were removed and cultivated at 60% humidity. Symptom development was observed for 30 days. The isolates, Xhc JJ2001, produced infection and necrosis in inoculated carrot leaves (Fig. 1C).
The isolates, Xhc JJ2001, was confirmed as pathogenic and was used for sequencing. Genomic DNA was extracted from the JJ2001 strain using an MGTM genomic DNA purification kit (Epicenter, Madison, WI, USA). High-quality, high-molecular-weight genomic DNA (8 μg) was used to prepare a 20 kb SMRTbell template for PacBioRSII sequencing. Genomic DNA size was determined using a Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). A final 10 μl library was prepared using a PacBio DNA Template Prep Kit 1.0. SMRTbell templates were annealed using the PacBio DNA/Polymerase Binding Kit P6. For sequencing, PacBio DNA Sequencing Kit 4.0 and eight SMRT cells were used, and each SMRT cell was captured using a PacBio RS-II (Pacific Biosciences, Menlo Park, CA, USA) sequencing platform from Macrogen (Seoul, Korea). Subreads generated by PacBio RS-II were assembled using the Hierarchical Genome Assembly Process (HGAP) (Chin et al., 2013) with default options. The chromosomal and plasmid assembly of the Xhc JJ2001 PacBio sequence was honed with additional Illumina short read sequencing.
For Illumina sequencing, genomic DNA integrity was confirmed by agarose gel electrophoresis and DNA was quantified using Quant-IT PicoGreen (Invitrogen, Carlsbad, CA, USA). Sequencing libraries were prepared with a TruSeq DNA Nano Sample Preparation Kit (Illumina, Inc., San Diego, CA, USA). Purified libraries were quantified using qPCR according to the manufacturer’s qPCR Quantification Protocol Guide (KAPA Library Quantification Kit for Illumina Sequencing Platform) and validated using an Agilent Technologies 2200 TapeStation. Libraries were then sequenced using HiSeq (Illumina, Inc.). Illumina raw data was processed to remove adapters and filtered by quality for error correction. Trimmomatic was used for adapter trimming. For error correction, reads with a base quality of at least 90% Q20 or higher were used. Genome assembly was modified using high-quality HiseqXten reads from Pilon v1.21 (Walker et al., 2014).
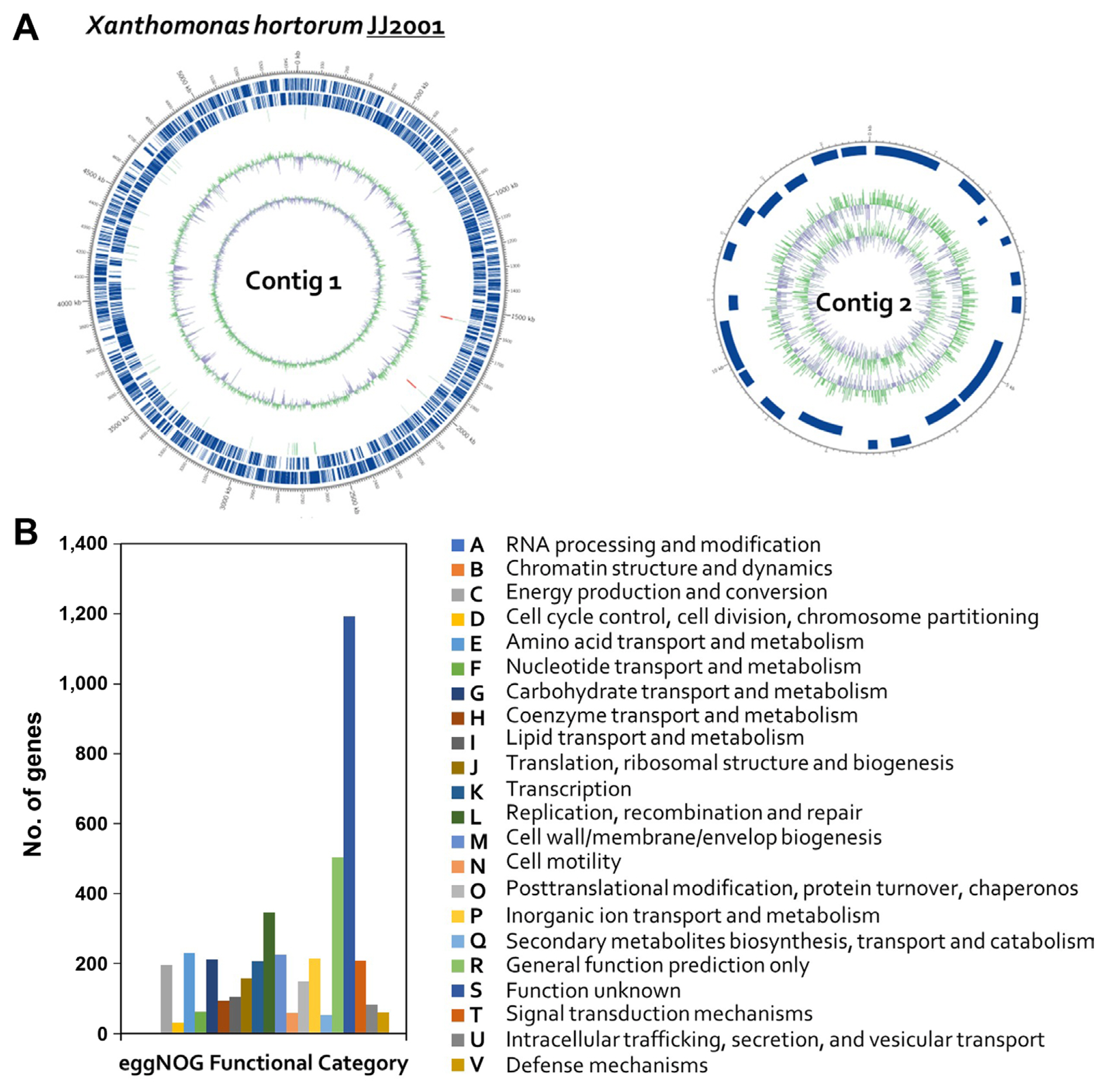
After assembly, Illumina reads were used to improve genomic sequence accuracy with Pilon. Subreads were mapped to assembled contigs to generate consensus sequences at depth-range. By adjusting the contigs, more accurate nucleotide genome sequences could be obtained and adapted for different analysis protocols (Bioto lnc., Daejeon, Korea). Genome annotation was performed using a prokaryotic genome annotation pipeline (Tatusova et al. 2016), including transfer RNAs and ribosomal RNAs. Prokka v1.13 (Seemann, 2014) was used for gene prediction and default annotations with the following options: --compliant, --rnamer, and --addgenes. For further annotation, predicted protein sets were identified with InterProScan v5.30-69.0 (Jones et al., 2014) and psiblast v2.4.0 (Camacho et al., 2009) using EggNOG DB v4.5 (Huerta Cepas et al., 2016). Circos v0.69.3 (Krzywinski et al., 2009) was used to generate a circular map representing each contig.
The draft genome sequence of Xhc JJ2001 had a total length of 5,458,083 bp and an N50 value of 5,443,372 bp. Contig 1 was 5,443,372 bp long and had a G + C content of 63.63 mol%. Contig 2 was 14,711 bp long and had a G + C content of 62.66 mol% (Table 1, Fig. 2A). The genome contained 4,571 coding sequence, six rRNA gene operons, and 54 tRNA genes. Genomic features are shown in Fig. 2. Using the EggNOG v. 4.5 database, 4,391 genes were classified into clusters of orthologous genes (COGs) functional groups. The most abundant COGs category was “Function unknown” (S; 1,193 genes), followed by “Replication, recombination and repair” (L; 346 genes), “Amino acid transport and metabolism” (E; 230 genes), “Cell wall, membrane biosynthesis” (M; 226 genes), “Inorganic ion transport and metabolism” (P; 215 genes), “Carbohydrate transport and metabolism” (G; 212 genes), “Signal transduction mechanisms” (T; 209 genes), and “Transcription” (K; 207 genes) noted as major categories (Fig. 2B). To evaluate similarities between the isolated pathogenic Xhc JJ2001 genome and genomes of other Xanthomonas species, the assembled M081 Xhc genome sequence was downloaded from the NCBI database, as well as sequences from the NCBI Xanthomonas RefSeq database (1,660 sequences), and species were compared by calculation of distance values (Ondov et al., 2016). The gene sequence-based species identification was performed using protein sequence analysis with the BLAST v2.9.0 program (Zhang et al., 2000) against the NCBI NR bacteria database (275,186,762 sequences), with blast (e-value ≤ 1e-10, best-match) used to estimate species via organism information for matching genes.
Comparative genome analysis was performed based on Mash-distance using genome data from Xhc JJ2001 and other Xanthomonas genome data from NCBI. Mash-distance was the lowest for Xhc CFBP7900 (GenBank accession no. NZ_CAJDKC010000000) 0.00892796, and second for Xhc M081, 0.0090861. Together with the results of the pathogenicity analysis, these results confirmed that Xhc JJ2001 was a X. hortorum pv. carotae (Supplementary Table 3).
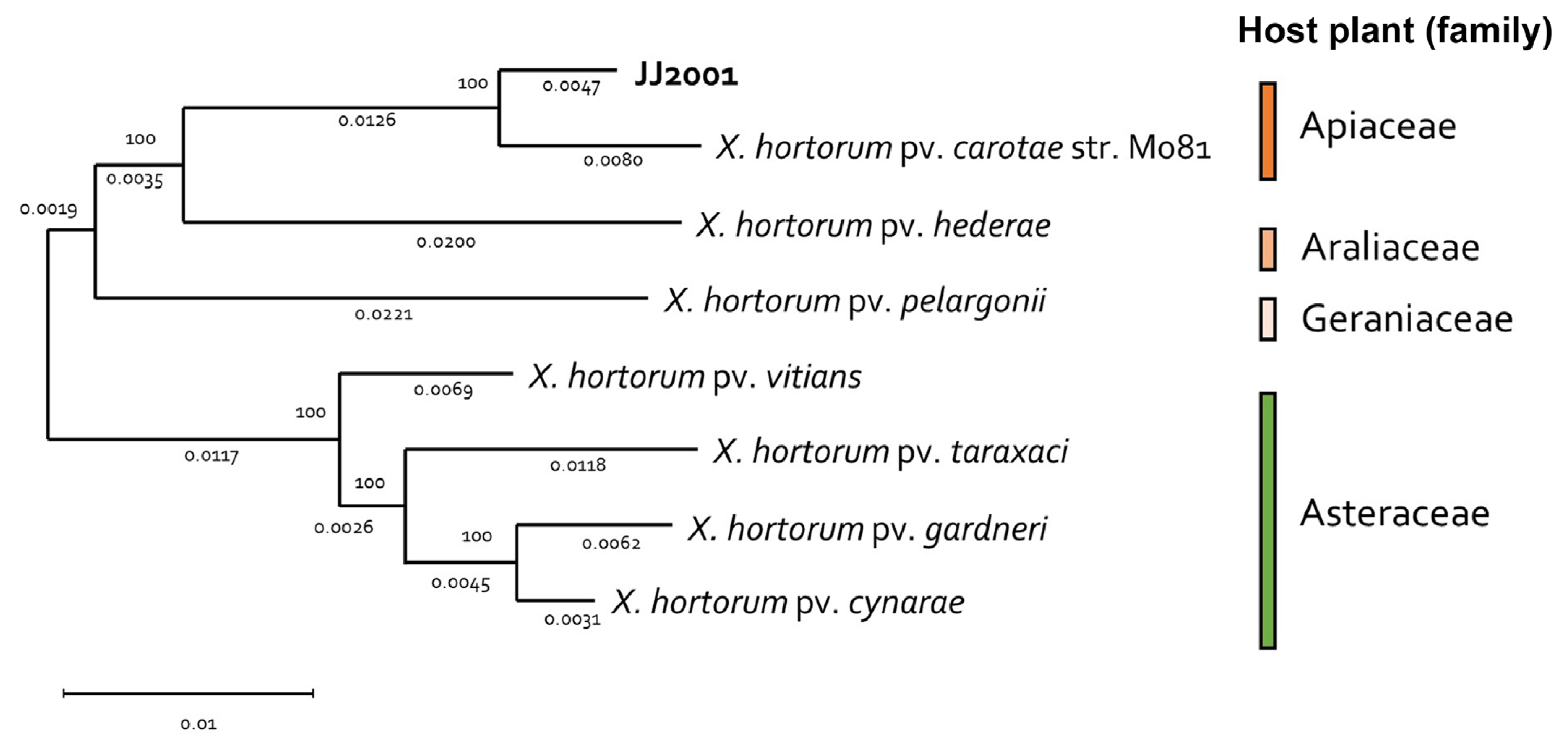
In addition, 7 X. hortorum pathovar were selected and NCBI genome data (GCF_000505565.1, GCF_028580375.1, GCF_028370135.1, GCF_014338485.1, GCF_021352955.1, GCF_001908755.1, and GCF_021352995.1) was received, the genome was compared and analyzed. In the case of the isolated strain, JJ2001, it was found to have the most similar genome to Xhc (GCF_00505565.1), just like the MLSA results. In addition, in the case of each Pathovar, it was found to form a group similarly according to the host plant family (Fig. 3). Overall, there was no significant difference in gene size and G/C content between isolates and other pathovars (Table 2). Furthermore, through Mauve analysis, differences in pathogenic gene islands for each pathovar of X. hortoum were compared and analyzed. In the case of 7 pathovars, there was a difference in gene direction, but the distribution of pathogenic genes was the same (Fig. 4). The pathogenic gene island of JJ2001 was divided into two clusters, hrcT, hrpB7, hrcN, hrpB, hrpB4, hrcJ, jrpB2, and hrpB1 share a promoter, and hrcU and hrcV form another group. In the case of JJ2001, it had 7 avirulence protein encoding genes and 17 genes related to the type secretion system in the genome including the above genes (Supplementary Table 4). The complete genome sequence of X. hotorum JJ2001 has been deposited in NCBI GenBank assembly accession number CP101417 (https://www.ncbi.nlm.nih.gov/nuccore/CP101417.1/).
In conclusion, this study, hybrid assembly of long and contiguous Pacbio reads with short Illumina reads obtained from the novel JJ2001 Xhc strain generated a complete, high-resolution genome. Additional complete genomic assembly of X. hortorum pathovar could facilitate the exhaustive investigation of nucleotide and structural variations across Xanthomonas strains with complex taxonomic systems, which will aid in understanding the Xhc pathogen and provide effective and sustainable management strategies.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement1
Supplement1 Print
Print